
 Submit Manuscript
Submit Manuscript
Review Article | Open Access
The role of cardiovascular aging in heart failure
Denise Greco1, Kofi Oti Boakye-Yiadom2
1Department of Physiology, Faculty of Science, Charles University, Prague, Czech Republic.
2Department of Pharmaceutics, School of Pharmacy, Kwame Nkrumah University of Science and Technology, Kumasi, Ghana.
Correspondence: Kofi Oti Boakye-Yiadom (Department of Pharmaceutics, School of Pharmacy, Kwame Nkrumah University of Science and Technology, Kumasi, Ghana; E-mail: otiboakye1000@gmail.com).
Asia-Pacific Journal of Surgical & Experimental Pathology 2025, 2: 9-18. https://doi.org/10.32948/ajsep.2025.01.10
Received: 01 Nov 2024 | Accepted: 20 Jan 2025 | Published online: 29 Jan 2025
Key words cardiovascular aging, heart failure, genetics, metabolism, inflammation
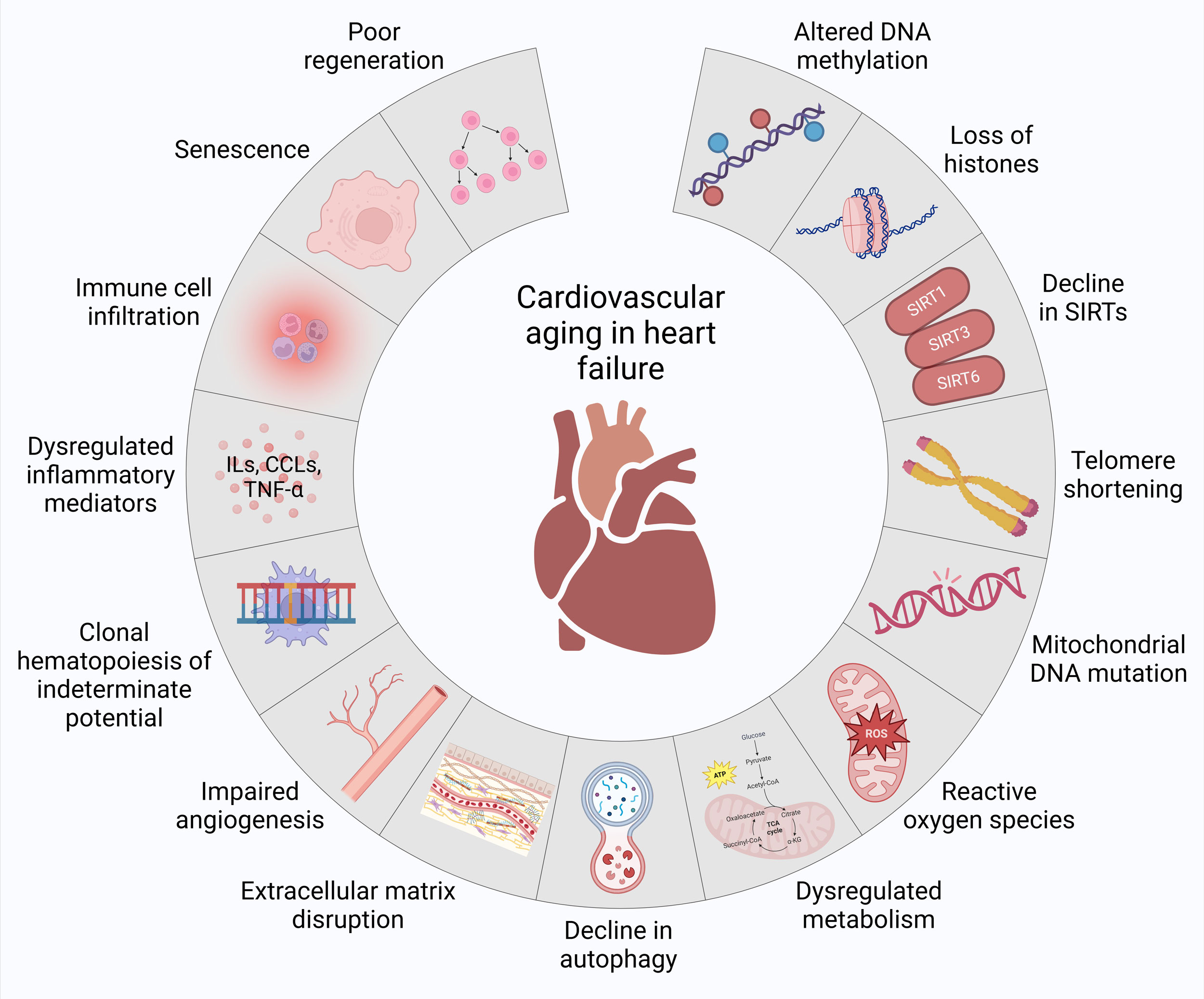 Figure 1. Molecular basis of cardiovascular aging in heart failure. At molecular level, the increased vulnerability to heart failure is linked to altered DNA methylation, loss of histones, decline in sirtuins (SIRTs), telomere shortening, mitochondrial DNA mutations, reactive oxygen species, dysregulated metabolism, decline in autophagy, extracellular matrix disruption, impaired angiogenesis, clonal hematopoiesis of indeterminate potential, dysregulated inflammatory mediators, immune cell infiltration, senescence and poor regeneration during cardiovascular aging.
Figure 1. Molecular basis of cardiovascular aging in heart failure. At molecular level, the increased vulnerability to heart failure is linked to altered DNA methylation, loss of histones, decline in sirtuins (SIRTs), telomere shortening, mitochondrial DNA mutations, reactive oxygen species, dysregulated metabolism, decline in autophagy, extracellular matrix disruption, impaired angiogenesis, clonal hematopoiesis of indeterminate potential, dysregulated inflammatory mediators, immune cell infiltration, senescence and poor regeneration during cardiovascular aging.
|
Table 1. Molecular basis of cardiovascular aging in heart failure. |
|||
|
Factor |
Genes/Proteins/Pathways |
Impact on heart failure |
Ref |
|
Altered DNA methylation |
DUX4, DNMTs, METTL3 |
Impairs cardiac cell viability, promotes dysfunction, and exacerbates heart failure through epigenetic changes |
[11-13] |
|
Loss of histones |
HDAC1, HDAC2, HDAC3, HDAC5, HDAC9 |
Promotes cardiac hypertrophy, dysfunction, and aging-related characteristics |
[15-17] |
|
Decline in SIRTs |
SIRT1, SIRT3, SIRT6 |
Increases oxidative stress and mitochondrial dysfunction, worsening cardiac performance and reducing lifespan |
[18-24] |
|
Telomere Shortening |
TERC, TRF2, Tert, p21, Chk2 |
Accelerates cardiomyocyte senescence, apoptosis, and aging-related cardiac dysfunction |
[26-30] |
|
Mitochondrial DNA mutations |
Polg, Pgc-1α, Twinkle (Twnk), p66Shc |
Disrupts mitochondrial biogenesis and contributes to oxidative stress, fibrosis, and arrhythmias |
[31-37] |
|
Reactive oxygen species |
NOX, NLRP3, eNOS, NF-κB, mCAT |
Drives oxidative stress, DNA damage, and chronic inflammation, promoting heart failure progression |
[38-44] |
|
Dysregulated metabolism |
IRS, AMPK, PPARs, mTOR |
Increases lipotoxicity, reduces metabolic flexibility, and exacerbates mitochondrial dysfunction in cardiomyocytes |
[45-49] |
|
Decline in autophagy |
mTOR, SIRT1, Lamp2, Atg5, Atg7, Becn1, Akt, Gsk3α, HSPB6, miR-22 |
Drives accumulation of damaged organelles, sarcomere disarray, and impaired cardiac function |
[51-61] |
|
Extracellular matrix disruption |
MMPs, TIMPs, YAP, TAZ |
Induces fibrosis, arterial stiffness, and increased cardiac workload, driving heart failure progression |
[62-66] |
|
Impaired angiogenesis |
VEGF, eNOS, PKG, NO |
Limits oxygen and nutrient delivery, worsening cardiac performance and ischemic injury |
[67-70] |
|
Clonal hematopoiesis |
DNMT3A, TET2, IL-6 |
Elevates systemic inflammation, promoting cardiac fibrosis, hypertrophy, and dysfunction |
[72-76] |
|
Dysregulated inflammatory mediators |
NF-κB, IL-1α, IL-1β, IL-6, IL-17, TNF-α, MCP1, CXCL1, CCR2 |
Enhances chronic inflammation, cardiomyocyte hypertrophy, and contractile dysfunction |
[77-85] |
|
Immune cell infiltration |
Neutrophils, monocytes, macrophages, T-cells |
Sustains inflammation, fibrosis, and maladaptive cardiac remodeling |
[88-94] |
|
Senescence |
p53, p16INK4a, Tgf-β2, Gdf15, Edn3 |
Promotes hypertrophy, mitochondrial dysfunction, and left ventricular diastolic dysfunction |
|
|
Poor regeneration |
Enhanced cell cycle progression-driven polyploidy |
Reduces cardiomyocyte renewal, impairing regenerative capacity and leading to cardiomyopathy |
[106-110] |
Loss of histones
Modifications to histones after translation, such as acetylation and deacetylation, as well as methylation and demethylation, are linked to cardiac dysfunction related to aging and associated diseases [14]. The loss of histone deacetylases (HDACs) such as HDAC1, 2, 3, 5, and 9 has been associated with the promotion of aging-related characteristics, including cardiac hypertrophy, dysfunction, increased susceptibility to heart injury, and shortened lifespan [15-17]. These findings highlight the significant impact of histones on cardiac dysfunction and heart failure.
Decline in SIRTs
Reduced expression of SIRT1 in cardiomyocytes from patients with advanced heart failure and animal models correlates with increased oxidative stress, inflammation, and apoptosis [18, 19]. SIRT1 mitigates oxidative stress in cardiomyocytes by regulating proteins such as manganese superoxide dismutase (MnSOD), thioredoxin1 (TRX1), and Bcl-xL, and helps prevent cardiomyocyte apoptosis via the NF-κB p65/miR-155/brain-derived neurotrophic factor (BDNF) signaling pathway, providing protection against heart failure in rats [20]. A lack of SIRT3 may impair mitochondrial function in the heart and worsen heart failure as organisms age. Additionally, SIRT3 plays a role in endothelial metabolism and angiogenesis, influencing the onset and progression of heart failure. Deletion of SIRT3 specifically in endothelial cells disrupts glucose transport, reduces glucose utilization in cardiomyocytes, and heightens susceptibility to pressure overload-induced heart failure in vivo [21, 22]. Similarly, SIRT6 has demonstrated protective effects in heart failure, with its expression reduced in patients with chronic heart failure and in animal models. Overexpression of SIRT6 improves survival rates in heart failure mice, possibly through the upregulation of telomerase [23, 24]. These findings highlight the significant impact of SIRTs on cardiovascular dysfunction and heart failure.
Telomere shortening
Telomere length has emerged as a significant biomarker of cellular aging [25]. Mice with TERC gene deficiency undergo significant telomere shortening, leading to increased p21-mediated cell cycle arrest in cardiomyocytes, culminating in cardiac aging and dysfunction [26, 27]. In mice, partial aortic constriction reduces the expression of telomeric repeat-binding factor 2 (Trf2), a shelterin protein, which accelerates telomere shortening and induces cardiomyocyte apoptosis, while overexpressing Tert or Trf2 alleviates these effects [28]. Although telomere length in human myocardium decreases with age [29] and correlations between shortened telomeres, reduced TRF2 levels, and increased apoptosis in end-stage heart failure patients have been observed [28], contradictory findings suggest that telomere attrition in heart failure may not reflect typical cardiac aging, as it is associated with substantial DNA damage in cardiomyocytes [30]. These findings highlight the significant impact of telomere shortening on cardiovascular dysfunction and heart failure.
Mitochondrial DNA mutations
Elevated mitochondrial DNA mutations undermine the integrity of mitochondria, disrupting mitochondrial biogenesis and leading to an increased production of reactive oxygen species [31]. The "Mutator" mice, which carry a homologous mutation in the mitochondrial polymerase gamma (Polgm/m), exhibit impaired mitochondrial function, and the development of early aging traits, including cardiac hypertrophy, dilated cardiomyopathy, and fibrosis, leading to an average lifespan of just 12 months [32, 33]. When these Polgm/m mice are crossed with antioxidant catalase (mCAT) overexpressing mice, there is a partial restoration of the cardiac aging and heart failure phenotypes [34]. Deletion of Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (Pgc-1α), a crucial regulator of mitochondrial biogenesis, results in cardiac impairment as early as 7 to 8 months of age in mice [35]. Overexpression of the myocardial Twinkle (Twnk) helicase in mice accelerates mitochondrial DNA deletions, contributing to the development of arrhythmias as they age [36]. Mutations in the p66Shc gene, which modulates reactive oxygen species production, result in reduced mitochondrial reactive oxygen species, increased resistance to reactive oxygen species-induced apoptosis, and a longer lifespan [37]. These findings highlight the significant impact of mitochondrial mutations on cardiovascular dysfunction and heart failure.
Reactive oxygen species
Mitochondrial reactive oxygen species may impair the mitochondrial respiratory chain, leading to oxidative stress, DNA and protein damage, lipid peroxidation, and the opening of the mitochondrial permeability transition pore (MPTP). This cascade triggers the release of cytochrome C, which initiates chronic proteome alterations and apoptosis, contributing to acute cardiovascular events and heart failure [38]. Additionally, mitochondrial reactive oxygen species have been implicated in the activation of the NLRP3 inflammasome and the induction of cardiomyocyte pyroptosis in dilated cardiomyopathy, highlighting a novel mechanism in heart failure onset and progression [39]. Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX) is a major source of reactive oxygen species, promoting lipid peroxidation of the mitochondrial membrane and activation of redox-sensitive mitochondrial potassium channels, which further generates mitochondrial reactive oxygen species from the electron transport chain [40]. The overexpression of NOX intensifies oxidative stress in aging blood vessels. This elevated oxidative environment, in turn, activates inflammatory pathways such as NF-κB [41]. Impaired nitric oxide (NO) signaling manifests endothelial dysfunction, an increase in oxidative stress, and persistent inflammation. Reduced endothelial NO synthase (eNOS) activity leads to lower NO availability, a situation worsened by high levels of reactive oxygen species, especially superoxide anions, which combine with NO to form peroxynitrite, thereby reducing NO’s ability to induce vasodilation in smooth muscle cells [42]. In contrast, mitochondrial-specific overexpression of the antioxidant mCAT has been shown to extend lifespan by mitigating cardiac aging and reducing oxidative damage to mitochondrial DNA and proteins [43, 44]. These findings highlight the significant impact of reactive oxygen species on cardiovascular dysfunction and heart failure.
Dysregulated metabolism
In the aging heart, insulin resistance impairs the ability of cells to uptake glucose by disrupting glucose transporter activity, resulting in an increased reliance on fatty acids for energy production. This shift toward fatty acid metabolism undermines metabolic flexibility, leading to mitochondrial dysfunction and an increase in the generation of reactive oxygen species [45]. Alterations in the signaling pathways associated with insulin receptor substrates (IRS) and the AMP-activated protein kinase (AMPK) are critical contributors to this metabolic dysregulation [46]. Insulin resistance may lead to elevated levels of circulating free fatty acids (FFAs), which promote lipid accumulation within cardiomyocytes. This accumulation, due to diminished lipid oxidation, induces lipotoxicity, further compromising cardiac function [47, 48]. Additionally, age-related disruptions in the metabolic signaling pathways, such as peroxisome proliferator-activated receptors (PPARs) signaling pathway and the activation of the mechanistic target of rapamycin (mTOR) pathway exacerbate cardiac hypertrophy, fibrosis, and defective autophagic processes, which are hallmark features of heart failure [49]. These findings highlight the significant impact of metabolic dysregulation on cardiovascular dysfunction and heart failure.
In aging hearts, there is a marked upregulation of senescence markers such as p53 and p16Ink4a, contributing to pathological changes like hypertrophy, mitochondrial dysfunction, increased cardiomyocyte death, reduced contractility, and heart failure [95, 96]. Senescent cells accumulate progressively over time, exerting harmful paracrine effects on surrounding cells and systemic consequences on distant tissues via senescence-associated secretory phenotype (SASP) [97]. Recent studies indicate that these senescent cells play a crucial role in cardiac remodeling and dysfunction with aging [95, 98]. In addition to the traditional proinflammatory SASP factors—IL-1α, IL-1β, IL-6, and TNF-α—which promote localized and systemic inflammation, emerging research highlights a wider array of secreted proteins and RNAs contributing to aging-related diseases such as heart failure [7, 99]. Cardiomyocytes in senescence can induce similar changes in neighboring cells by secreting nontraditional SASP factors, such as endothelin 3 (Edn3), transforming growth factor beta 2 (Tgf-β2), and growth differentiation factor 15 (Gdf15), as revealed by studies on cardiomyocytes isolated from old mice [100]. Age-related cellular senescence induces an inflammatory phenotype in both vascular and myocardial endothelial cells, as seen in the hearts of senescence-accelerated mouse models. These changes contribute to diastolic dysfunction and left ventricular hypertrophy, which are often present in heart failure [101]. These findings highlight the significant impact of senescence and SASP on cardiovascular dysfunction and heart failure.
Poor regeneration
Following cardiac injury, heart failure models show an increase in cell cycle activity [102], resembling the regenerative potential seen in neonatal cardiomyocytes, which can replicate and aid in cardiac repair [103, 104]. However, as cardiomyocytes mature postnatally, they undergo significant changes that progressively reduce their ability to proliferate [105]. Despite this, the enhanced cell cycle activity in damaged hearts typically leads to polyploidy rather than effective cardiomyocyte regeneration [106]. Adult cardiomyocytes exhibit a renewal rate ranging from 0.5% to 2% annually, suggesting a modest, albeit restricted and regenerative ability within the heart [107, 108]. This regenerative capacity primarily arises from the replication of existing cardiomyocytes rather than differentiation of stem cells [109]. However, this renewal process diminishes with advancing age, reflecting a reduced capacity to replace lost cardiomyocytes. This is particularly significant because even small-scale, experimentally induced loss of cardiomyocytes can lead to cardiomyopathy and mortality [110]. These findings highlight the significant impact of poor regeneration on cardiovascular dysfunction and heart failure.
No applicable.
Ethics approval
No applicable.
Data availability
The data will be available upon request.
Funding
None.
Authors’ contribution
DG and KOBY contributed to the conception, design, writing of this review article, drawing figures, make data table and submitted the final version of the manuscript.
Competing interests
None.
- Roger VL: Epidemiology of Heart Failure: A Contemporary Perspective. Circ Res 2021, 128(10): 1421-1434.
- Arrigo M, Jessup M, Mullens W, Reza N, Shah AM, Sliwa K, Mebazaa A: Acute heart failure. Nat Rev Dis Primers 2020, 6(1): 16.
- Benjamin EJ, Muntner P, Alonso A, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Chang AR, Cheng S, Das SR, et al: Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation 2019, 139(10): e56-e528.
- Strait JB, Lakatta EG: Aging-associated cardiovascular changes and their relationship to heart failure. Heart Fail Clin 2012, 8(1): 143-164.
- Triposkiadis F, Xanthopoulos A, Butler J: Cardiovascular Aging and Heart Failure: JACC Review Topic of the Week. J Am Coll Cardiol 2019, 74(6): 804-813.
- Ya J, Bayraktutan U: Vascular Ageing: Mechanisms, Risk Factors, and Treatment Strategies. Int J Mol Sci 2023, 24(14): 11538.
- Li H, Hastings MH, Rhee J, Trager LE, Roh JD, Rosenzweig A: Targeting Age-Related Pathways in Heart Failure. Circ Res 2020, 126(4): 533-551.
- Franceschi C, Garagnani P, Parini P, Giuliani C, Santoro A: Inflammaging: a new immune-metabolic viewpoint for age-related diseases. Nat Rev Endocrinol 2018, 14(10): 576-590.
- Wang W, Zhang Y, Wang R, Shrestha Y, Xu Y, Peng L, Zhang J, Li J, Zhang L: Risk Factors And Epigenetic Markers Of Left Ventricular Diastolic Dysfunction With Preserved Ejection Fraction In A Community-Based Elderly Chinese Population. Clin Interv Aging 2019, 14: 1719-1728.
- Gilsbach R, Preissl S, Grüning BA, Schnick T, Burger L, Benes V, Würch A, Bönisch U, Günther S, Backofen R, et al: Dynamic DNA methylation orchestrates cardiomyocyte development, maturation and disease. Nat Commun 2014, 5: 5288.
- Movassagh M, Choy MK, Knowles DA, Cordeddu L, Haider S, Down T, Siggens L, Vujic A, Simeoni I, Penkett C, et al: Distinct epigenomic features in end-stage failing human hearts. Circulation 2011, 124(22): 2411-2422.
- Xiao D, Dasgupta C, Chen M, Zhang K, Buchholz J, Xu Z, Zhang L: Inhibition of DNA methylation reverses norepinephrine-induced cardiac hypertrophy in rats. Cardiovasc Res 2014, 101(3): 373-382.
- Dorn LE, Lasman L, Chen J, Xu X, Hund TJ, Medvedovic M, Hanna JH, van Berlo JH, Accornero F: The N(6)-Methyladenosine mRNA Methylase METTL3 Controls Cardiac Homeostasis and Hypertrophy. Circulation 2019, 139(4): 533-545.
- Papait R, Cattaneo P, Kunderfranco P, Greco C, Carullo P, Guffanti A, Viganò V, Stirparo GG, Latronico MV, Hasenfuss G, et al: Genome-wide analysis of histone marks identifying an epigenetic signature of promoters and enhancers underlying cardiac hypertrophy. Proc Natl Acad Sci U S A 2013, 110(50): 20164-20169.
- Herman AB, Occean JR, Sen P: Epigenetic dysregulation in cardiovascular aging and disease. J Cardiovasc Aging 2021, 1: 10.
- Montgomery RL, Potthoff MJ, Haberland M, Qi X, Matsuzaki S, Humphries KM, Richardson JA, Bassel-Duby R, Olson EN: Maintenance of cardiac energy metabolism by histone deacetylase 3 in mice. J Clin Invest 2008, 118(11): 3588-3597.
- Montgomery RL, Davis CA, Potthoff MJ, Haberland M, Fielitz J, Qi X, Hill JA, Richardson JA, Olson EN: Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev 2007, 21(14): 1790-1802.
- Lu TM, Tsai JY, Chen YC, Huang CY, Hsu HL, Weng CF, Shih CC, Hsu CP: Downregulation of Sirt1 as aging change in advanced heart failure. J Biomed Sci 2014, 21(1): 57.
- Wu YX, Xu RY, Jiang L, Chen XY, Xiao XJ: MicroRNA-30a-5p Promotes Chronic Heart Failure in Rats by Targeting Sirtuin-1 to Activate the Nuclear Factor-κB/NOD-Like Receptor 3 Signaling Pathway. Cardiovasc Drugs Ther 2023, 37(6): 1065-1076.
- Lin B, Zhao H, Li L, Zhang Z, Jiang N, Yang X, Zhang T, Lian B, Liu Y, Zhang C, et al: Sirt1 improves heart failure through modulating the NF-κB p65/microRNA-155/BNDF signaling cascade. Aging (Albany NY) 2020, 13(10): 14482-14498.
- Benigni A, Cassis P, Conti S, Perico L, Corna D, Cerullo D, Zentilin L, Zoja C, Perna A, Lionetti V, et al: Sirt3 Deficiency Shortens Life Span and Impairs Cardiac Mitochondrial Function Rescued by Opa1 Gene Transfer. Antioxid Redox Signal 2019, 31(17): 1255-1271.
- Zeng H, He X, Chen JX: Endothelial Sirtuin 3 Dictates Glucose Transport to Cardiomyocyte and Sensitizes Pressure Overload-Induced Heart Failure. J Am Heart Assoc 2020, 9(11): e015895.
- Saiyang X, Deng W, Qizhu T: Sirtuin 6: A potential therapeutic target for cardiovascular diseases. Pharmacol Res 2021, 163: 105214.
- Li Y, Meng X, Wang W, Liu F, Hao Z, Yang Y, Zhao J, Yin W, Xu L, Zhao R, et al: Cardioprotective Effects of SIRT6 in a Mouse Model of Transverse Aortic Constriction-Induced Heart Failure. Front Physiol 2017, 8: 394.
- López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G: Hallmarks of aging: An expanding universe. Cell 2023, 186(2): 243-278.
- Aix E, Gutiérrez-Gutiérrez Ó, Sánchez-Ferrer C, Aguado T, Flores I: Postnatal telomere dysfunction induces cardiomyocyte cell-cycle arrest through p21 activation. J Cell Biol 2016, 213(5): 571-583.
- Wong LS, Oeseburg H, de Boer RA, van Gilst WH, van Veldhuisen DJ, van der Harst P: Telomere biology in cardiovascular disease: the TERC-/- mouse as a model for heart failure and ageing. Cardiovasc Res 2009, 81(2): 244-252.
- Oh H, Wang SC, Prahash A, Sano M, Moravec CS, Taffet GE, Michael LH, Youker KA, Entman ML, Schneider MD: Telomere attrition and Chk2 activation in human heart failure. Proc Natl Acad Sci U S A 2003, 100(9): 5378-5383.
- Terai M, Izumiyama-Shimomura N, Aida J, Ishikawa N, Sawabe M, Arai T, Fujiwara M, Ishii A, Nakamura K, Takubo K: Association of telomere shortening in myocardium with heart weight gain and cause of death. Sci Rep 2013, 3: 2401.
- Sharifi-Sanjani M, Oyster NM, Tichy ED, Bedi KC, Jr., Harel O, Margulies KB, Mourkioti F: Cardiomyocyte-Specific Telomere Shortening is a Distinct Signature of Heart Failure in Humans. J Am Heart Assoc 2017, 6(9): e005086.
- Wu C, Zhang Z, Zhang W, Liu X: Mitochondrial dysfunction and mitochondrial therapies in heart failure. Pharmacol Res 2022, 175: 106038.
- Vermulst M, Wanagat J, Kujoth GC, Bielas JH, Rabinovitch PS, Prolla TA, Loeb LA: DNA deletions and clonal mutations drive premature aging in mitochondrial mutator mice. Nat Genet 2008, 40(4): 392-394.
- Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly YM, Gidlöf S, Oldfors A, Wibom R, et al: Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 2004, 429(6990): 417-423.
- Dai DF, Chen T, Wanagat J, Laflamme M, Marcinek DJ, Emond MJ, Ngo CP, Prolla TA, Rabinovitch PS: Age-dependent cardiomyopathy in mitochondrial mutator mice is attenuated by overexpression of catalase targeted to mitochondria. Aging Cell 2010, 9(4): 536-544.
- Arany Z, He H, Lin J, Hoyer K, Handschin C, Toka O, Ahmad F, Matsui T, Chin S, Wu PH, et al: Transcriptional coactivator PGC-1 alpha controls the energy state and contractile function of cardiac muscle. Cell Metab 2005, 1(4): 259-271.
- Baris OR, Ederer S, Neuhaus JF, von Kleist-Retzow JC, Wunderlich CM, Pal M, Wunderlich FT, Peeva V, Zsurka G, Kunz WS, et al: Mosaic Deficiency in Mitochondrial Oxidative Metabolism Promotes Cardiac Arrhythmia during Aging. Cell Metab 2015, 21(5): 667-677.
- Migliaccio E, Giorgio M, Mele S, Pelicci G, Reboldi P, Pandolfi PP, Lanfrancone L, Pelicci PG: The p66shc adaptor protein controls oxidative stress response and life span in mammals. Nature 1999, 402(6759): 309-313.
- Dey S, DeMazumder D, Sidor A, Foster DB, O'Rourke B: Mitochondrial ROS Drive Sudden Cardiac Death and Chronic Proteome Remodeling in Heart Failure. Circ Res 2018, 123(3): 356-371.
- Zeng C, Duan F, Hu J, Luo B, Huang B, Lou X, Sun X, Li H, Zhang X, Yin S, et al: NLRP3 inflammasome-mediated pyroptosis contributes to the pathogenesis of non-ischemic dilated cardiomyopathy. Redox Biol 2020, 34: 101523.
- Zhang Y, Murugesan P, Huang K, Cai H: NADPH oxidases and oxidase crosstalk in cardiovascular diseases: novel therapeutic targets. Nat Rev Cardiol 2020, 17(3): 170-194.
- Sena CM, Leandro A, Azul L, Seiça R, Perry G: Vascular Oxidative Stress: Impact and Therapeutic Approaches. Front Physiol 2018, 9: 1668.
- Janaszak-Jasiecka A, Płoska A, Wierońska JM, Dobrucki LW, Kalinowski L: Endothelial dysfunction due to eNOS uncoupling: molecular mechanisms as potential therapeutic targets. Cell Mol Biol Lett 2023, 28(1): 21.
- Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, Emond M, Coskun PE, Ladiges W, Wolf N, Van Remmen H, et al: Extension of murine life span by overexpression of catalase targeted to mitochondria. Science 2005, 308(5730): 1909-1911.
- Dai DF, Santana LF, Vermulst M, Tomazela DM, Emond MJ, MacCoss MJ, Gollahon K, Martin GM, Loeb LA, Ladiges WC, et al: Overexpression of catalase targeted to mitochondria attenuates murine cardiac aging. Circulation 2009, 119(21): 2789-2797.
- Bhashyam S, Parikh P, Bolukoglu H, Shannon AH, Porter JH, Shen YT, Shannon RP: Aging is associated with myocardial insulin resistance and mitochondrial dysfunction. Am J Physiol Heart Circ Physiol 2007, 293(5): H3063-3071.
- Lee WS, Kim J: Insulin-like growth factor-1 signaling in cardiac aging. Biochim Biophys Acta Mol Basis Dis 2018, 1864(5 Pt B): 1931-1938.
- Livshits G, Kalinkovich A: Inflammaging as a common ground for the development and maintenance of sarcopenia, obesity, cardiomyopathy and dysbiosis. Ageing Res Rev 2019, 56: 100980.
- Bertero E, Maack C: Metabolic remodelling in heart failure. Nat Rev Cardiol 2018, 15(8): 457-470.
- Duszka K, Gregor A, Guillou H, König J, Wahli W: Peroxisome Proliferator-Activated Receptors and Caloric Restriction-Common Pathways Affecting Metabolism, Health, and Longevity. Cells 2020, 9(7): 1708.
- Taneike M, Yamaguchi O, Nakai A, Hikoso S, Takeda T, Mizote I, Oka T, Tamai T, Oyabu J, Murakawa T, et al: Inhibition of autophagy in the heart induces age-related cardiomyopathy. Autophagy 2010, 6(5): 600-606.
- Maejima Y, Kyoi S, Zhai P, Liu T, Li H, Ivessa A, Sciarretta S, Del Re DP, Zablocki DK, Hsu CP, et al: Mst1 inhibits autophagy by promoting the interaction between Beclin1 and Bcl-2. Nat Med 2013, 19(11): 1478-1488.
- Sciarretta S, Zhai P, Shao D, Maejima Y, Robbins J, Volpe M, Condorelli G, Sadoshima J: Rheb is a critical regulator of autophagy during myocardial ischemia: pathophysiological implications in obesity and metabolic syndrome. Circulation 2012, 125(9): 1134-1146.
- Hariharan N, Maejima Y, Nakae J, Paik J, Depinho RA, Sadoshima J: Deacetylation of FoxO by Sirt1 Plays an Essential Role in Mediating Starvation-Induced Autophagy in Cardiac Myocytes. Circ Res 2010, 107(12): 1470-1482.
- Tanaka Y, Guhde G, Suter A, Eskelinen EL, Hartmann D, Lüllmann-Rauch R, Janssen PM, Blanz J, von Figura K, Saftig P: Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2-deficient mice. Nature 2000, 406(6798): 902-906.
- Wang F, He Q, Gao Z, Redington AN: Atg5 knockdown induces age-dependent cardiomyopathy which can be rescued by repeated remote ischemic conditioning. Basic Res Cardiol 2021, 116(1): 47.
- Li S, Liu C, Gu L, Wang L, Shang Y, Liu Q, Wan J, Shi J, Wang F, Xu Z, et al: Autophagy protects cardiomyocytes from the myocardial ischaemia-reperfusion injury through the clearance of CLP36. Open Biol 2016, 6(8): 160177.
- Zhou J, Freeman TA, Ahmad F, Shang X, Mangano E, Gao E, Farber J, Wang Y, Ma XL, Woodgett J, et al: GSK-3α is a central regulator of age-related pathologies in mice. J Clin Invest 2013, 123(4): 1821-1832.
- Hua Y, Zhang Y, Ceylan-Isik AF, Wold LE, Nunn JM, Ren J: Chronic Akt activation accentuates aging-induced cardiac hypertrophy and myocardial contractile dysfunction: role of autophagy. Basic Res Cardiol 2011, 106(6): 1173-1191.
- Liu GS, Zhu H, Cai WF, Wang X, Jiang M, Essandoh K, Vafiadaki E, Haghighi K, Lam CK, Gardner G, et al: Regulation of BECN1-mediated autophagy by HSPB6: Insights from a human HSPB6(S10F) mutant. Autophagy 2018, 14(1): 80-97.
- Zhu H, Tannous P, Johnstone JL, Kong Y, Shelton JM, Richardson JA, Le V, Levine B, Rothermel BA, Hill JA: Cardiac autophagy is a maladaptive response to hemodynamic stress. J Clin Invest 2007, 117(7): 1782-1793.
- Gupta SK, Foinquinos A, Thum S, Remke J, Zimmer K, Bauters C, de Groote P, Boon RA, de Windt LJ, Preissl S, et al: Preclinical Development of a MicroRNA-Based Therapy for Elderly Patients With Myocardial Infarction. J Am Coll Cardiol 2016, 68(14): 1557-1571.
- Bassat E, Mutlak YE, Genzelinakh A, Shadrin IY, Baruch Umansky K, Yifa O, Kain D, Rajchman D, Leach J, Riabov Bassat D, et al: The extracellular matrix protein agrin promotes heart regeneration in mice. Nature 2017, 547(7662): 179-184.
- Morikawa Y, Heallen T, Leach J, Xiao Y, Martin JF: Dystrophin-glycoprotein complex sequesters Yap to inhibit cardiomyocyte proliferation. Nature 2017, 547(7662): 227-231.
- Cabral-Pacheco GA, Garza-Veloz I, Castruita-De la Rosa C, Ramirez-Acuña JM, Perez-Romero BA, Guerrero-Rodriguez JF, Martinez-Avila N, Martinez-Fierro ML: The Roles of Matrix Metalloproteinases and Their Inhibitors in Human Diseases. Int J Mol Sci 2020, 21(24): 9739.
- Wang L, Luo JY, Li B, Tian XY, Chen LJ, Huang Y, Liu J, Deng D, Lau CW, Wan S, et al: Integrin-YAP/TAZ-JNK cascade mediates atheroprotective effect of unidirectional shear flow. Nature 2016, 540(7634): 579-582.
- Feola M: The influence of arterial stiffness in heart failure: a clinical review. J Geriatr Cardiol 2021, 18(2): 135-140.
- Pérez-Gutiérrez L, Ferrara N: Biology and therapeutic targeting of vascular endothelial growth factor A. Nat Rev Mol Cell Biol 2023, 24(11): 816-834.
- Gogiraju R, Bochenek ML, Schäfer K: Angiogenic Endothelial Cell Signaling in Cardiac Hypertrophy and Heart Failure. Front Cardiovasc Med 2019, 6: 20.
- Layland J, Li JM, Shah AM: Role of cyclic GMP-dependent protein kinase in the contractile response to exogenous nitric oxide in rat cardiac myocytes. J Physiol 2002, 540(Pt 2): 457-467.
- Cornuault L, Rouault P, Duplàa C, Couffinhal T, Renault MA: Endothelial Dysfunction in Heart Failure With Preserved Ejection Fraction: What are the Experimental Proofs? Front Physiol 2022, 13: 906272.
- Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, Lindsley RC, Mermel CH, Burtt N, Chavez A, et al: Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 2014, 371(26): 2488-2498.
- Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, McConkey M, Gupta N, Gabriel S, Ardissino D, et al: Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N Engl J Med 2017, 377(2): 111-121.
- Xie M, Lu C, Wang J, McLellan MD, Johnson KJ, Wendl MC, McMichael JF, Schmidt HK, Yellapantula V, Miller CA, et al: Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med 2014, 20(12): 1472-1478.
- Sano S, Oshima K, Wang Y, Katanasaka Y, Sano M, Walsh K: CRISPR-Mediated Gene Editing to Assess the Roles of Tet2 and Dnmt3a in Clonal Hematopoiesis and Cardiovascular Disease. Circ Res 2018, 123(3): 335-341.
- Pascual-Figal DA, Bayes-Genis A, Díez-Díez M, Hernández-Vicente Á, Vázquez-Andrés D, de la Barrera J, Vazquez E, Quintas A, Zuriaga MA, Asensio-López MC, et al: Clonal Hematopoiesis and Risk of Progression of Heart Failure With Reduced Left Ventricular Ejection Fraction. J Am Coll Cardiol 2021, 77(14): 1747-1759.
- Wang Y, Sano S, Yura Y, Ke Z, Sano M, Oshima K, Ogawa H, Horitani K, Min KD, Miura-Yura E, et al: Tet2-mediated clonal hematopoiesis in nonconditioned mice accelerates age-associated cardiac dysfunction. JCI Insight 2020, 5(6): e135204.
- Fiordelisi A, Iaccarino G, Morisco C, Coscioni E, Sorriento D: NFkappaB is a Key Player in the Crosstalk between Inflammation and Cardiovascular Diseases. Int J Mol Sci 2019, 20(7): 1599.
- Ninh VK, Brown JH: The contribution of the cardiomyocyte to tissue inflammation in cardiomyopathies. Curr Opin Physiol 2021, 19: 129-134.
- Birks EJ, Felkin LE, Banner NR, Khaghani A, Barton PJ, Yacoub MH: Increased toll-like receptor 4 in the myocardium of patients requiring left ventricular assist devices. J Heart Lung Transplant 2004, 23(2): 228-235.
- Van Tassell BW, Raleigh JM, Abbate A: Targeting interleukin-1 in heart failure and inflammatory heart disease. Curr Heart Fail Rep 2015, 12(1): 33-41.
- Van Tassell BW, Seropian IM, Toldo S, Mezzaroma E, Abbate A: Interleukin-1β induces a reversible cardiomyopathy in the mouse. Inflamm Res 2013, 62(7): 637-640.
- Jia X, Buckley L, Sun C, Al Rifai M, Yu B, Nambi V, Virani SS, Selvin E, Matsushita K, Hoogeveen RC, et al: Association of interleukin-6 and interleukin-18 with cardiovascular disease in older adults: Atherosclerosis Risk in Communities study. Eur J Prev Cardiol 2023, 30(16): 1731-1740.
- Wang Y, Zhu S, Wei W, Tu Y, Chen C, Song J, Li J, Wang C, Xu Z, Sun S: Interleukin-6 knockout reverses macrophage differentiation imbalance and alleviates cardiac dysfunction in aging mice. Aging (Albany NY) 2020, 12(20): 20184-20197.
- Moro-García MA, Echeverría A, Galán-Artímez MC, Suárez-García FM, Solano-Jaurrieta JJ, Avanzas-Fernández P, Díaz-Molina B, Lambert JL, López-Larrea C, Morís de la Tassa C, et al: Immunosenescence and inflammation characterize chronic heart failure patients with more advanced disease. Int J Cardiol 2014, 174(3): 590-599.
- Papamichail A, Kourek C, Briasoulis A, Xanthopoulos A, Tsougos E, Farmakis D, Paraskevaidis I: Targeting Key Inflammatory Mechanisms Underlying Heart Failure: A Comprehensive Review. Int J Mol Sci 2023, 25(1): 510.
- Hu D, Cui YX, Wu MY, Li L, Su LN, Lian Z, Chen H: Cytosolic DNA sensor cGAS plays an essential pathogenetic role in pressure overload-induced heart failure. Am J Physiol Heart Circ Physiol 2020, 318(6): H1525-h1537.
- Epelman S, Lavine KJ, Beaudin AE, Sojka DK, Carrero JA, Calderon B, Brija T, Gautier EL, Ivanov S, Satpathy AT, et al: Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 2014, 40(1): 91-104.
- Ma Y: Role of Neutrophils in Cardiac Injury and Repair Following Myocardial Infarction. Cells 2021, 10(7): 1676.
- Vagnozzi RJ, Maillet M, Sargent MA, Khalil H, Johansen AKZ, Schwanekamp JA, York AJ, Huang V, Nahrendorf M, Sadayappan S, et al: An acute immune response underlies the benefit of cardiac stem cell therapy. Nature 2020, 577(7790): 405-409.
- Frantz S, Falcao-Pires I, Balligand JL, Bauersachs J, Brutsaert D, Ciccarelli M, Dawson D, de Windt LJ, Giacca M, Hamdani N, et al: The innate immune system in chronic cardiomyopathy: a European Society of Cardiology (ESC) scientific statement from the Working Group on Myocardial Function of the ESC. Eur J Heart Fail 2018, 20(3): 445-459.
- Aoyagi T, Matsui T: The Cardiomyocyte as a Source of Cytokines in Cardiac Injury. J Cell Sci Ther 2011, 2012(S5): 003.
- Zhang L, Yang X, Jiang G, Yu Y, Wu J, Su Y, Sun A, Zou Y, Jiang H, Ge J: HMGB1 enhances mechanical stress-induced cardiomyocyte hypertrophy in vitro via the RAGE/ERK1/2 signaling pathway. Int J Mol Med 2019, 44(3): 885-892.
- Patel B, Bansal SS, Ismahil MA, Hamid T, Rokosh G, Mack M, Prabhu SD: CCR2(+) Monocyte-Derived Infiltrating Macrophages Are Required for Adverse Cardiac Remodeling During Pressure Overload. JACC Basic Transl Sci 2018, 3(2): 230-244.
- Delgobo M, Heinrichs M, Hapke N, Ashour D, Appel M, Srivastava M, Heckel T, Spyridopoulos I, Hofmann U, Frantz S, et al: Terminally Differentiated CD4(+) T Cells Promote Myocardial Inflammaging. Front Immunol 2021, 12: 584538.
- Chen MS, Lee RT, Garbern JC: Senescence mechanisms and targets in the heart. Cardiovasc Res 2022, 118(5): 1173-1187.
- Leri A, Franco S, Zacheo A, Barlucchi L, Chimenti S, Limana F, Nadal-Ginard B, Kajstura J, Anversa P, Blasco MA: Ablation of telomerase and telomere loss leads to cardiac dilatation and heart failure associated with p53 upregulation. Embo j 2003, 22(1): 131-139.
- Chaib S, Tchkonia T, Kirkland JL: Cellular senescence and senolytics: the path to the clinic. Nat Med 2022, 28(8): 1556-1568.
- Wang M, Shah AM: Age-associated pro-inflammatory remodeling and functional phenotype in the heart and large arteries. J Mol Cell Cardiol 2015, 83: 101-111.
- Roh JD, Hobson R, Chaudhari V, Quintero P, Yeri A, Benson M, Xiao C, Zlotoff D, Bezzerides V, Houstis N, et al: Activin type II receptor signaling in cardiac aging and heart failure. Sci Transl Med 2019, 11(482): eaau8680.
- Anderson R, Lagnado A, Maggiorani D, Walaszczyk A, Dookun E, Chapman J, Birch J, Salmonowicz H, Ogrodnik M, Jurk D, et al: Length-independent telomere damage drives post-mitotic cardiomyocyte senescence. Embo j 2019, 38(5): e100492.
- Gevaert AB, Shakeri H, Leloup AJ, Van Hove CE, De Meyer GRY, Vrints CJ, Lemmens K, Van Craenenbroeck EM: Endothelial Senescence Contributes to Heart Failure With Preserved Ejection Fraction in an Aging Mouse Model. Circ Heart Fail 2017, 10(6): e003806.
- Vujic A, Lerchenmüller C, Wu TD, Guillermier C, Rabolli CP, Gonzalez E, Senyo SE, Liu X, Guerquin-Kern JL, Steinhauser ML et al: Exercise induces new cardiomyocyte generation in the adult mammalian heart. Nat Commun 2018, 9(1): 1659.
- Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, Olson EN, Sadek HA: Transient regenerative potential of the neonatal mouse heart. Science 2011, 331(6020): 1078-1080.
- Porrello ER, Mahmoud AI, Simpson E, Johnson BA, Grinsfelder D, Canseco D, Mammen PP, Rothermel BA, Olson EN, Sadek HA: Regulation of neonatal and adult mammalian heart regeneration by the miR-15 family. Proc Natl Acad Sci U S A 2013, 110(1): 187-192.
- Karbassi E, Fenix A, Marchiano S, Muraoka N, Nakamura K, Yang X, Murry CE: Cardiomyocyte maturation: advances in knowledge and implications for regenerative medicine. Nat Rev Cardiol 2020, 17(6): 341-359.
- Derks W, Murganti F, Bergmann O: Cardiomyocyte renewal in the failing heart: lessons from the neonate? Biophys Rev 2020, 12(4): 785-787.
- Senyo SE, Steinhauser ML, Pizzimenti CL, Yang VK, Cai L, Wang M, Wu TD, Guerquin-Kern JL, Lechene CP, Lee RT: Mammalian heart renewal by pre-existing cardiomyocytes. Nature 2013, 493(7432): 433-436.
- Bergmann O, Zdunek S, Felker A, Salehpour M, Alkass K, Bernard S, Sjostrom SL, Szewczykowska M, Jackowska T, Dos Remedios C, et al: Dynamics of Cell Generation and Turnover in the Human Heart. Cell 2015, 161(7): 1566-1575.
- Eschenhagen T, Bolli R, Braun T, Field LJ, Fleischmann BK, Frisén J, Giacca M, Hare JM, Houser S, Lee RT, et al: Cardiomyocyte Regeneration: A Consensus Statement. Circulation 2017, 136(7): 680-686.
- Mehdipour M, Park S, Huang GN: Unlocking cardiomyocyte renewal potential for myocardial regeneration therapy. J Mol Cell Cardiol 2023, 177: 9-20.
- Ashley EA: Towards precision medicine. Nat Rev Genet 2016, 17(9): 507-522.
- Ibrahim NE, Gaggin HK, Konstam MA, Januzzi JL, Jr.: Established and Emerging Roles of Biomarkers in Heart Failure Clinical Trials. Circ Heart Fail 2016, 9(9): e002528.
Asia-Pacific Journal of Surgical & Experimental Pathology
ISSN 2977-5817 (Online)
 Copyright © Asia Pac J Surg Exp & Pathol. This
work is licensed under a Creative Commons AttributionNonCommercial-No Derivatives 4.0 International (CC BY-NC-ND 4.0)
License.
Copyright © Asia Pac J Surg Exp & Pathol. This
work is licensed under a Creative Commons AttributionNonCommercial-No Derivatives 4.0 International (CC BY-NC-ND 4.0)
License.