
 Submit Manuscript
Submit Manuscript
Review Article | Open Access
CRISPR/Cas9 potential applications in cancer immunotherapy by gene-editing and immune checkpoint signaling pathway inhibition
Mehab Ishfaq1
1Barcelona Institute of Bioinformatics, The Universitat Politècnica de Catalunya, Barcelona, 08034, Spain.
Correspondence: Mehab Ishfaq (Barcelona Institute of Bioinformatics, The Universitat Politècnica de Catalunya, Barcelona, 08034, Spain; E-mail: mehabishfaqkhn786@gmail.com).
Asia-Pacific Journal of Surgical & Experimental Pathology 2024, 1: 94-102. https://doi.org/10.32948/ajsep.2024.12.25
Received: 27 Aug 2024 | Accepted: 23 Dec 2024 | Published online: 31 Dec 2024
Key words CRISPR-Cas9, cancer immunotherapy, CAR-T, immune checkpoint inhibition
Checkpoint blockade therapy has gained a lot of importance with the developments in immunology research, whereas this has demonstrated its worth clinically, especially in hematological malignancies, with CAR-T cell therapy [6]. CAR-T cell therapy does have its advantages despite having some disadvantages like insufficient and poor-quality T cell availability from the individual and the presence of infection and serious complications with donor-derived T cells. Consequently, researchers are focusing on strategies to create universal or off-the-shelf CAR-T cells. Safety concerns related to allogeneic CAR-T cell transfer stem from the presence of endogenous alpha-beta T-cell receptors (TCR) and human leukocyte antigen (HLA) molecules on donor T cells.
The CRISPR/Cas9 system is a genomic editing method that has lately attracted attention for developing effective owl phenotypes across various domestic animal species [7]. In Cas9, the single-guide RNA (sgRNA) employs this as a guide to cleave double-strand fragments of DNA into smaller predetermined loci [8]. Initially, Cas9 identifies a protospacer adjacent motif (PAM) site, and it is then guided to the DSB by a sgRNA. Consequently, this DSB can be repaired by either non-homologous end joining (NHEJ) or homologous-directed repair (HDR). While it makes accurate sequence corrections using an external repair template, NHEJ is generally led by insertions or deletions (InDels), which are often used in disrupting genes. There is no doubt about the simplicity, accuracy, and straightforward application of CRISPR/Cas9 as compared to previous genome-editing methods [9].
CRISPR/Cas9 technology has tremendous promise for the enhancement of immunotherapy against cancer, given its tremendously strong efficacy in gene editing. It permits high-throughput screening in identifying new therapeutic targets and biomarkers as well as genes related to treatment resistance [10] in immunotherapy. Moreover, effective modification of adenoviral genomes is possible through CRISPR technology. It may also bring novel immunotherapy approaches for cancer by knocking down immune checkpoint genes such as PD-1, PD-L1, and CTLA-4 via CRISPR/Cas9 techniques [11]. In addition, CRISPR/is also a way of improving the production of CAR-T cell therapies and creating allogeneic CAR-T cells without graft-versus-host disease (GVHD) by rupturing the TCR beta chains and beta-2-microglobulin (B2M), which are important parts of HLA-I molecules [12]. Furthermore, the CRISPR/Cas9 technique has been employed in enhancing tumor cell phagocytosis using macrophages by hindering signaling mechanisms.
On the other hand, earlier instruments like transcription activator-like effector nucleases (TALENs) and zinc-finger nucleases (ZFNs) used specially made proteins to recognize and bind target DNA sequences [15]. Since it is much easier to create unique CRISPR-guided RNAs than it is to synthesize the proteins needed for ZFN or TALEN systems, the use of RNA as a targeting mechanism proved revolutionary.
Like many biomedical breakthroughs, CRISPR has its roots in natural biological systems. Variants of CRISPR systems have been found in archaea and phages since they were first identified as a bacterial defensive mechanism against bacteriophages, which are viruses that infect bacteria [16]. Despite its initial purpose as a component of the bacterial immune system, scientists soon realized and modified CRISPR for its potential in genome editing. The two main parts of the system are a protein complex called Cas9, which cleaves double-stranded DNA as a nuclease, and a guide RNA (gRNA), which guides the editing machinery to the desired gene [17] (Figure 2).
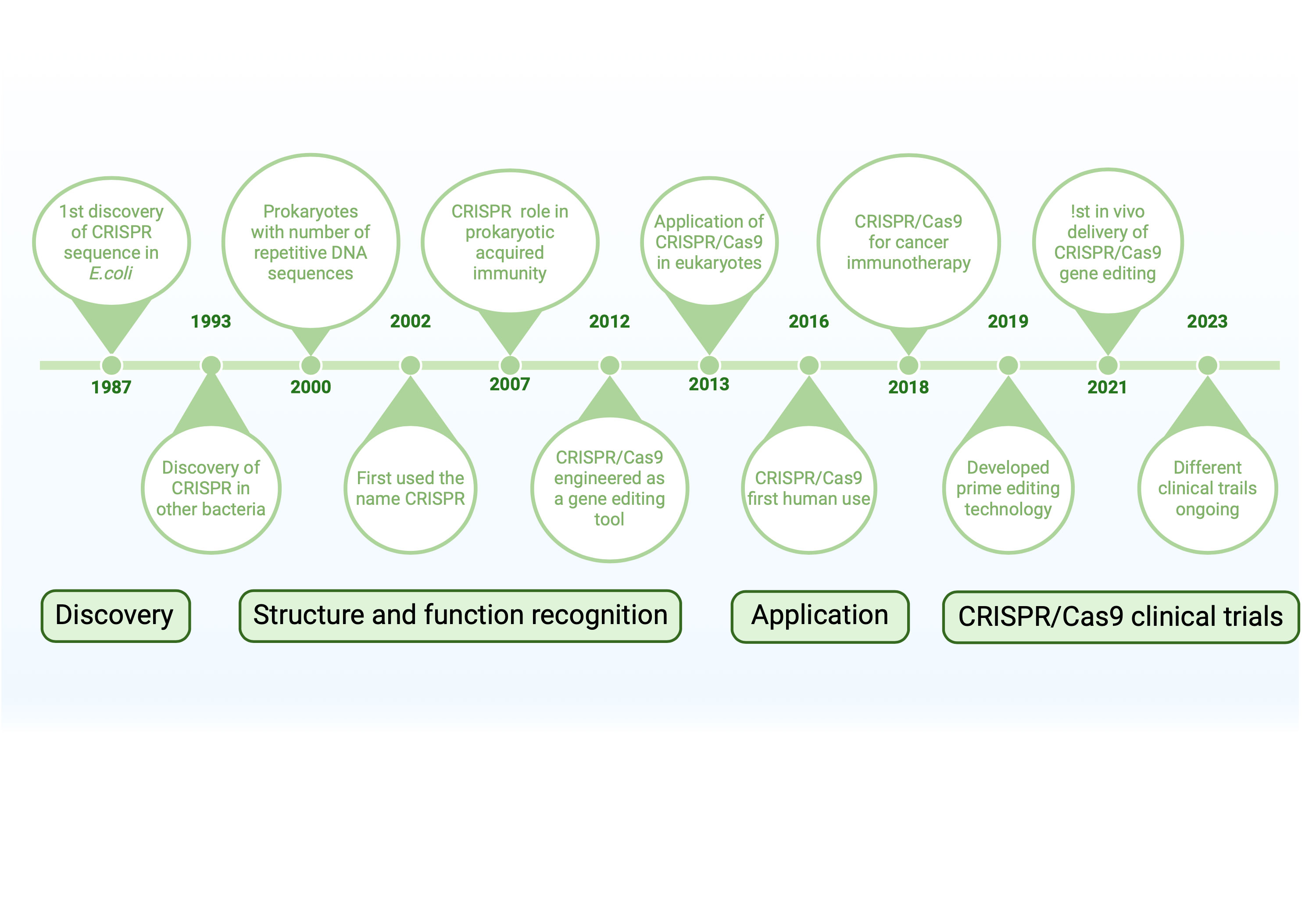 Figure 1. A timeline of significant turning points in the development and use of CRISPR/Cas9 technology is shown in this graphic. The discovery of CRISPR sequences in E. coli in 1987 marked the beginning of important developments, such as the identification of CRISPR's function in prokaryotic immunity in 2002, its use as a tool for gene editing in 2013, and its initial usage in humans in 2016. Current clinical studies (2023) and prime editing (2021) are examples of recent advancements. These developments are divided into four stages in the timeline: discovery, structural recognition, application, and clinical testing.
Figure 1. A timeline of significant turning points in the development and use of CRISPR/Cas9 technology is shown in this graphic. The discovery of CRISPR sequences in E. coli in 1987 marked the beginning of important developments, such as the identification of CRISPR's function in prokaryotic immunity in 2002, its use as a tool for gene editing in 2013, and its initial usage in humans in 2016. Current clinical studies (2023) and prime editing (2021) are examples of recent advancements. These developments are divided into four stages in the timeline: discovery, structural recognition, application, and clinical testing.
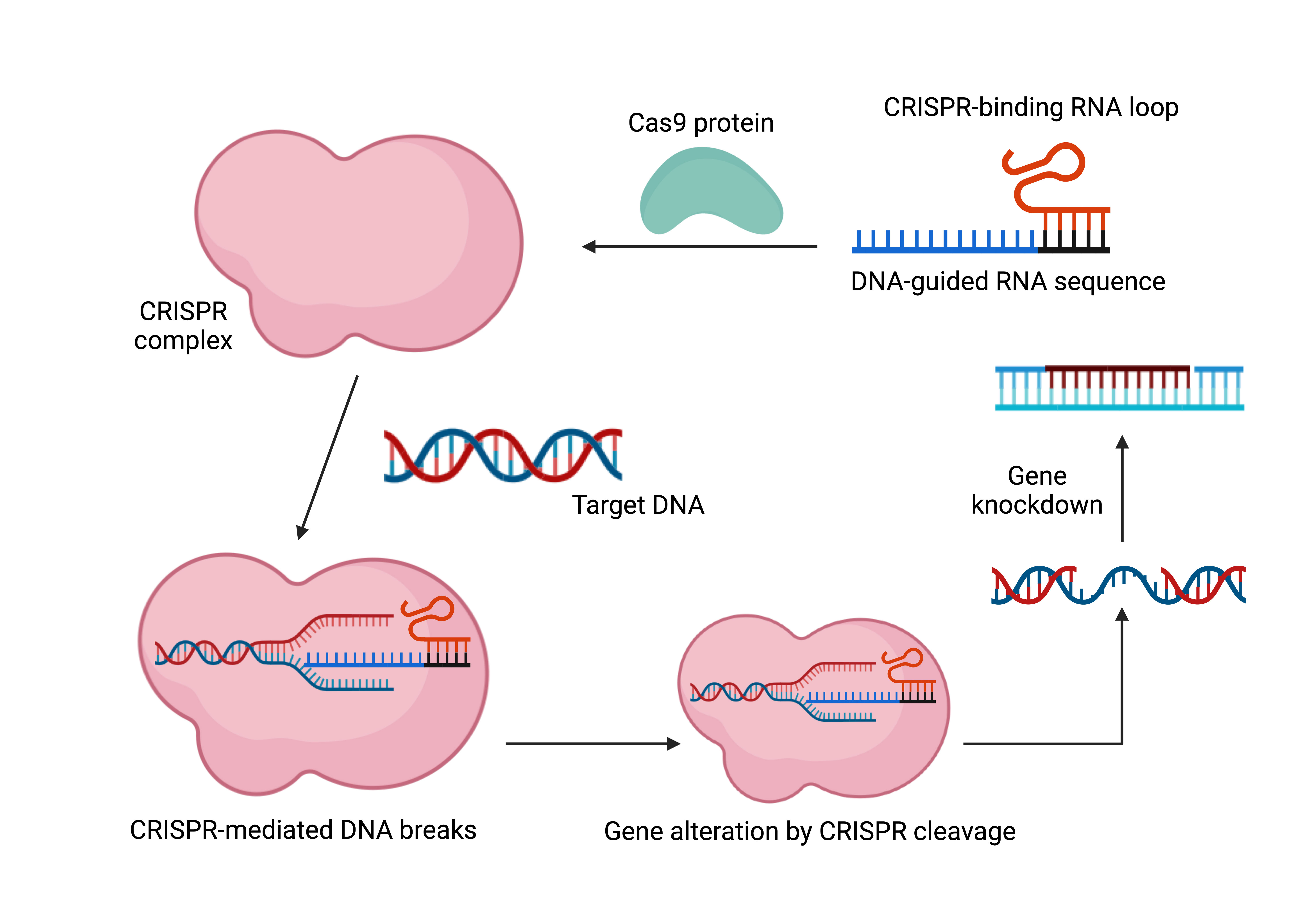 Figure 2. CRISPR complex development setup. A guide RNA (gRNA) directed at the desired gene binds to Cas9 in the CRISPR/Cas9 system to form the CRISPR complex. The CRISPR complex is formed when the gRNA containing the desired gene binds to Cas9. A loop area in the gRNA makes Cas9 binding easier. After then, the CRISPR complex looks for a loop region in the target gene that makes Cas9 binding easier. Next, the CRISPR complex targets double strand breaks in the target gene. If a donor template is available, gene knock-in may take place following breakage. If not, a repair mechanism will occur in the DNA sequence, eventually resulting in a gene that is not functioning.
Figure 2. CRISPR complex development setup. A guide RNA (gRNA) directed at the desired gene binds to Cas9 in the CRISPR/Cas9 system to form the CRISPR complex. The CRISPR complex is formed when the gRNA containing the desired gene binds to Cas9. A loop area in the gRNA makes Cas9 binding easier. After then, the CRISPR complex looks for a loop region in the target gene that makes Cas9 binding easier. Next, the CRISPR complex targets double strand breaks in the target gene. If a donor template is available, gene knock-in may take place following breakage. If not, a repair mechanism will occur in the DNA sequence, eventually resulting in a gene that is not functioning.
The U.S. Food and Drug Administration (FDA) has endorsed Kymriah and Yescarta, two anti-CD19 CAR-T cell therapies, for the treatment of pediatric and young adult B lymphoblastic leukemia (B-ALL) and adult diffuse large B-cell lymphoma (DLBCL), respectively, due to their remarkable clinical performance. The FDA endorsed the use of Tecartus in July 2020 for the treatment of adult patients with MCL. Breyanzi, the fourth CAR-T treatment, was also authorized in February 2021 for people with relapsed or refractory large B-cell lymphoma. There are now several registered clinical experiments underway, and new CAR-T structures and applications are being created quickly [19].
Despite these remarkable therapeutic developments, several obstacles prevent many patients from benefiting from T-cell therapy. First off, patients, particularly those with severe diseases, are unable to fully utilize this immunotherapy due to the time-consuming and expensive nature of CAR-T cell construction, which is individualized [20]. Second, it is frequently challenging to get sufficient high-quality T cells from individuals who are lymphopenic or in poor health during the production process. Even if enough immune cells are gathered, the manufacturing process cannot be successfully finished. Furthermore, there are hazards associated with T-cell production. In one instance, the CAR gene was inadvertently integrated into a single leukemic B cell, causing a B-cell leukemia patient to relapse nine months after acquiring anti-CD19 CAR-T cell therapy [21]. Finally, the clinical effects of autologous CAR-T products are diverse and unpredictable due to their heterogeneity.
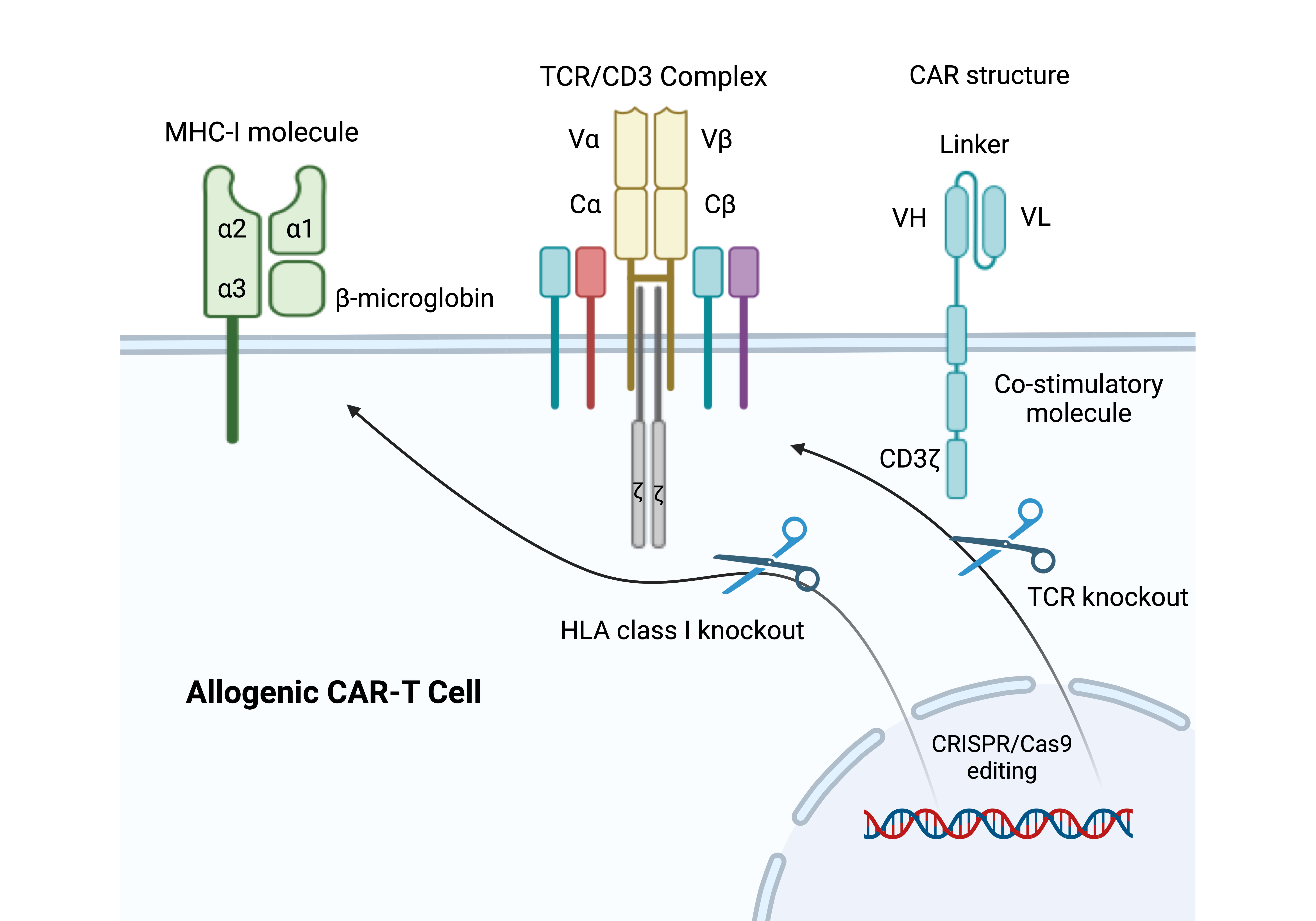 Figure 3. CRISPR/Cas9-Based Engineering of Universal CAR-T Cells. In this figure, the endogenous TCR locus and MHC-I molecules are knocked out in order to construct universal CAR-T cells using the CRISPR/Cas9 technology. By preventing immune rejection and graft-versus-host disease (GVHD), these changes make it possible to produce allogeneic CAR-T cells for more extensive therapeutic uses.
Figure 3. CRISPR/Cas9-Based Engineering of Universal CAR-T Cells. In this figure, the endogenous TCR locus and MHC-I molecules are knocked out in order to construct universal CAR-T cells using the CRISPR/Cas9 technology. By preventing immune rejection and graft-versus-host disease (GVHD), these changes make it possible to produce allogeneic CAR-T cells for more extensive therapeutic uses.
The effective delivery of perturbation reagents to target cells, which might differ substantially between organs, is crucial to the effectiveness of CRISPR-GEMMs. Because of this, the present uses of CRISPR-GEMMs are mostly restricted to liver, lung, and brain tumors [24]. Notwithstanding these drawbacks, organ-specific tumor models have enormous potential for pinpointing immune responses unique to tumors, opening the door for developments in precision immuno-oncology [25].
The functional particularization of effector T cells has been investigated using mouse infection models [26]. For example, in vivo, metabolic CRISPR screening was made possible by injecting Cas9-expressing CD4+ T cells into naïve mice infected with the lymphocytic choriomeningitis virus (LCMV). Using this method, ETNK1 and PCYT2 were found to be important regulators in deciding the fate of T follicular helper (TFH) cells as opposed to TH1 cells [27].
Using animal inflammatory models, it is possible to find genes that influence adaptive immune feedback in vivo [28]. In this experiment, sgRNA library-transduced CD4+ T cells were injected into Rag1-deficient mice that lack lymphocytes and had been vaccinated with ovalbumin (OVA) to induce lung inflammation. This led to the discovery of MTHFD2 as a metabolic gatekeeper that controls T-cell proliferation in the lungs and inflammatory reactions [29]. CRISPR/Cas9 has also been exploited to identify regulators of enhanced effector functions of CD8+ T cells in mouse models with tumors. For in vivo CRISPR screenings, the below mice implanted orthotopically with either GL261 glioblastoma or E0771-OVA triple-negative breast cancer were used. From these screenings, DHX37, PDIA3, and MGAT5 were identified as negative regulators of T-cell responses [30].
The safety and viability of modifying T cells using CRISPR/Cas9 have been assessed in various clinical trials. Twelve patients with non-small cell lung cancer (NSCLC) got involved in a trial conducted by Lu and associates that used PD-1-edited T cells. In this study, patients' peripheral blood cells were isolated, and CRISPR/Cas9 was used in a lab setting to knock out the PDCD1 gene. Patients were then given fresh injections of the altered cells. Instead of focusing on the treatment's effectiveness, the trial's main goal was to evaluate its safety and possible adverse effects.
Another CRISPR clinical trial in 2022 revealed poor editing efficiency, with just a median of 6% of T cells effectively modified, along with moderate adverse effects such as fever, rash, and exhaustion. Even yet, after two months, 11 patients still had detectable levels of modified T cells, albeit at low levels. These results imply that the strategy is initially safe, workable, and has tolerable side effects [33]. Stadtmauer's research in 2020 showed that, upon therapy, altered T cells could effectively infiltrate and engraft within tumor areas. With negative reactions falling within an acceptable range, this offered more proof of safety [34]. Similarly, in later phase I trials, Wang and colleagues [36] gave PD-1 gene-edited mesothelin-specific CAR T cells to patients with solid tumors, and Lacey and Fraietta [35] implanted edited T cells into patients with advanced NSCLC. The findings of these investigations were all in agreement: Gene editing in immune cells using CRISPR/Cas9 is safe, practical, and well-tolerated in clinical settings.
|
Table 1. CRISPR’s potential applications. |
|||
|
Cancer Type |
Target Genes |
CRISPR Method |
Reference |
|
Breast cancer |
FASN |
Knockdown |
[62] |
|
Breast cancer |
PARP1 |
Genome screening of novel pathways |
[63] |
|
Prostate cancer |
ERβ |
Genome screening of novel pathways |
[64] |
|
Prostate cancer |
TP53 |
sgRNA and Cas9-fused adenine base editor |
[65] |
|
Bladder cancer |
CTLA-4
|
Evaluate gene phenotypes via knockdown |
[66] |
|
Colon cancer |
PKC |
sgRNA and Cas9-fused adenine base editor |
[67] |
|
Colorectal |
KRAS, BRAF |
Genome screening of novel pathways |
[68] |
|
Intestinal tumors |
Colorectal cancer driver genes |
Evaluate gene phenotypes via knockdown |
[69] |
|
Glioblastoma |
TERT |
sgRNA and Cas9-fused adenine base editor |
[70] |
|
Lung metastases |
Genes on non-metastatic cancer cell line |
Evaluate gene phenotypes via knockdown |
[71] |
|
Melanoma
|
Novel gene involved in PD-1 resistance |
Evaluate gene phenotypes via knockdown |
[72] |
|
Table 2. CRISPR in clinical practice. |
|||
|
Name/Trial |
Status |
Disease |
Reference |
|
Lovotibeglogene autotemcel |
Currently evaluated in clinical trials |
SCD, Thalassemia, TDT |
[73] |
|
CLIMB THAL-111, CLIMB SCD-111 |
Currently evaluated in clinical trials |
SCD, TDT |
[74] |
|
Voretigene neparvovec |
2017 FDA approved |
Retinal dystrophy |
[75] |
|
Onasemnogene abeparvovec |
2019 FDA approved |
SMA (Pediatric Patients, < 2 y/o) |
[76] |
Exhausted tumor-infiltrating T lymphocytes have been shown to express higher levels of inhibitory receptors, including CTLA-4, CD152, PD-1, CD279, LAG3, IM-3, HAVCR2, CD244, CD160, TIGIT, and several others, according to gene expression analyses and phenotypic studies in both humans and mice [39]. These immune checkpoint markers have attracted a lot of consideration in cancer research lately because of their crucial functions in controlling anti-tumor immunity.
CD28 is an essential co-stimulatory protein that enhances TCR signaling and activates T lymphocytes by binding to CD80 (B7-1) or CD86 (B7-2) in response to antigen identification by the TCR/CD3 complex [40]. But only CD4+ and CD8+ T cells express CTLA-4, a homolog of CD28, which binds to the same ligands as CD28 to mediate inhibitory signals [41]. By increasing ligand availability to CD28, blocking CTLA-4 with mAbs or other genetic techniques improves T cell activation. To increase the immune system's capacity to combat tumor cells, ipilimumab, a recombinant human IgG1 monoclonal antibody that targets CTLA-4, was created [42]. When compared against the gp100 vaccination alone, clinical trials showed that ipilimumab, either alone or in conjunction with the gp100 peptide vaccine, remarkably enhanced comprehensive survival in patients with metastatic melanoma [42]. Consequently, in March 2011, the FDA in the United States authorized ipilimumab for the treatment of metastatic melanoma. Since then, anti-CTLA-4 treatments have been broadly studied for different malignancies, such as prostate, breast, and NSCLC [43].
To disrupt CTLA-4, CRISPR/Cas9 technology has been used in addition to monoclonal antibody treatments. CRISPR/Cas9 was used by Shi et al. (2017) and Zhang et al. (2019) to eliminate CTLA-4 from CTLs. According to their research, CTLA-4 deletion increased TNF-α and IFN-γ secretion in comparison to control groups, which improved anti-tumor efficacy [44, 45]. T cell activity is mostly controlled by the PD-1/PD-L1 immune checkpoint axis, especially when T cell receptors (TCRs) interact with MHC-peptide complexes that are presented by antigen-presenting cells (APCs) [46]. While its ligands, PD-L1, and PD-L2, are expressed on tumor cells and APCs, PD-1 is known to be expressed on a diversity of stimulated immune cells, such as T cells, monocytes, and dendritic cells [47]. The immune system's potential to fight malignancies is restricted by the interplay between PD-1 and PD-L1, which inhibits T cell activation and leads to T cell exhaustion [47]. Anti-PD-1 antibodies like nivolumab and pembrolizumab have shown impressive anti-tumor effects in cancers like melanoma, NSCLC, head and neck squamous cell carcinoma, and metastatic urothelial carcinoma [48]. Blocking this pathway with mAbs has been shown to restore T cell function.
Disrupting inhibitory genes in the PD-1/PD-L1 pathway has become a viable tactic as an alternative to antibody-based treatments. Peripheral blood T cells, CAR-T cells, and antigen-specific CTLs are among the T cell subsets whose PD-1 expression has been knocked out by recent developments using CRISPR/Cas9 technology [49]. Su et al. (2017) demonstrated that using electroporation to deliver plasmids encoding the CRISPR/Cas9 system could effectively knock out PD-1 in T cells without compromising their viability in vitro. Additionally, this modification enhanced T cell anti-tumor activity and cytokine secretion, particularly IFN-γ [50].
The safety and viability of PD-1 knockdown in therapeutic contexts are being investigated in clinical trials. For example, CRISPR/Cas9-modified T cells without PD-1 were safe but had little therapeutic efficacy in a completed clinical trial including patients with NSCLC [51]. While studies into the use of PD-1 knockout T cells for muscle-invasive bladder cancer, castration-resistant prostate cancer, and renal cell carcinoma were registered but subsequently withdrawn, additional trials assessing their use for advanced esophageal cancer (NCT03081715) have also been finished.
Targeting PD-L1 in tumor cells can enhance immunotherapy results in addition to T cells. For instance, Tu et al. created novel nanoparticles that were sensitive to mild acidity and included paclitaxel (PTX) and CRISPR/Cas9-Cdk5 plasmids. While the CRISPR/Cas9-Cdk5 system decreased PD-L1 expression on tumor cells, boosting anti-tumor immune responses, the PTX component caused immunogenic cell death and decreased the suppressive immune cell population [52]. A photosensitive CRISPR/Cas9 system was also developed by Zhao et al. (2020) to eliminate PD-L1. This technique effectively targeted PD-L1 in bulk cancer cells and cancer stem-like cells when activated by light, providing a new way to improve cancer immunotherapy [53].
For applications requiring prolonged CRISPR activity, viral vectors are often employed to deliver the CRISPR components at the DNA level. This strategy offers high transduction efficiency and enables precise control via promoter regulation, utilizing the host cell's transcriptional machinery. However, different viral systems present distinct limitations. Adeno-associated viruses (AAVs), for instance, can only accommodate payloads of approximately 4.5-5 kilobases, barely sufficient to include spCas9 and sgRNA, which together measure around 4.2 kilobases. While lentiviruses and adenoviruses can carry larger genetic cargo, they present other risks, such as unintended genomic integration and the induction of strong immune responses, which pose significant safety concerns [55].
Physical delivery methods, such as microinjection, electroporation, and hydrodynamic delivery, bypass the need for intermediary carriers. However, their efficiency in delivering CRISPR components in vivo is limited, often resulting in suboptimal targeting of cells [56]. Among non-viral delivery approaches, lipid nanoparticles have emerged as a promising option due to their long-term stability and compatibility with the immune system. Recent advancements have introduced additional non-viral systems for CRISPR delivery, including lipoplexes, polyplexes, cell-penetrating peptides, DNA nanoclews, and methods like induced transduction via osmocytosis and propanebetaine. Gold nanoparticles have also been explored, offering unique properties for gene editing studies [57].
In addition to the aforementioned challenges, CRISPR-related concerns also include the risk of runaway immune responses and spatiotemporal dysfunctions, which can lead to off-target effects and reduced productivity. To address these issues, researchers have developed engineered or split Cas enzymes that provide better control over the in vivo activity of the CRISPR-Cas system [58].
However, beyond these technical challenges, ethical considerations remain paramount. While the urgent need for more effective and tolerable therapies to combat the growing burden of malignancies is clear, the widespread use of CRISPR technology mustn't precede the resolution of the ethical dilemmas it raises. Failure to carefully address these concerns could lead to disastrous consequences [59]. Ethical issues such as the unintentional or intentional editing of tumor suppressor proteins like P53, as well as alterations to human somatic or germline cells, highlight the gravity of these debates [60]. Furthermore, irresponsible applications of CRISPR, like the case of CRISPR-modified twins in China, must be avoided to prevent a repeat of the setbacks experienced with other technologies, such as vaccines. Ensuring responsible and ethical use of CRISPR is essential for its continued development and safe application [61].
No applicable.
Ethics approval
No applicable.
Data availability
The data will be available upon request.
Funding
None.
Authors’ contribution
MI contributed to the conception, design, writing of this review article, drawing figures and submitted the final version of the manuscript.
Competing interests
None.
- Torre LA, Bray F, Siegel RL, Ferlay J, Lortet‐Tieulent J, Jemal A: Global cancer statistics, 2012. CA Cancer J Clin 2015, 65(2): 87-108.
- Stupp R, Hegi ME, Mason WP, Van Den Bent MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B, Belanger K et al: Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol 2009, 10(5): 459-466.
- Galon J, Bruni D: Tumor immunology and tumor evolution: intertwined histories. Immunity 2020, 52(1): 55-81.
- Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, Lu S, Kemberling H, Wilt C, Luber BS et al: Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357(6349): 409-413.
- Senzer NN, Kaufman HL, Amatruda T, Nemunaitis M, Reid T, Daniels G, Gonzalez R, Glaspy J, Whitman E, Harrington K et al: Phase II clinical trial of a granulocyte-macrophage colony-stimulating factor–encoding, second-generation oncolytic herpesvirus in patients with unresectable metastatic melanoma. J Clin Oncol 2009, 27(34): 5763-5771.
- Dai H, Wang Y, Lu X, Han W: Chimeric antigen receptors modified T-cells for cancer therapy. J Natl Cancer Inst 2016, 108(7): djv439.
- Rosenblum D, Gutkin A, Kedmi R, Ramishetti S, Veiga N, Jacobi AM, Schubert MS, Friedmann-Morvinski D, Cohen ZR, Behlke MA et al: CRISPR-Cas9 genome editing using targeted lipid nanoparticles for cancer therapy. Sci Adv 2020, 6(47): eabc9450.
- Jiang F, Doudna JA: CRISPR–Cas9 structures and mechanisms. Annu Rev Biophys 2017, 46(1): 505-529.
- Li J, Røise JJ, He M, Das R, Murthy N: Non-viral strategies for delivering genome editing enzymes. Adv Drug Deliv Rev 2021, 168: 99-117.
- Patel SJ, Sanjana NE, Kishton RJ, Eidizadeh A, Vodnala SK, Cam M, Gartner JJ, Jia L, Steinberg SM, Yamamoto TN et al: Identification of essential genes for cancer immunotherapy. Nature 2017, 548(7669): 537-542.
- Wieder T, Eigentler T, Brenner E, Röcken M: Immune checkpoint blockade therapy. J Allergy Clin Immunol 2018, 142(5): 1403-1414.
- Liu X, Zhao Y: CRISPR/Cas9 genome editing: Fueling the revolution in cancer immunotherapy. Curr Res Transl Med 2018, 66(2): 39-42.
- Ray M, Lee YW, Hardie J, Mout R, Yeşilbag Tonga G, Farkas ME, Rotello VM: CRISPRed macrophages for cell-based cancer immunotherapy. Bioconjug Chem 2018, 29(2): 445-450.
- Wang H, La Russa M, Qi LS: CRISPR/Cas9 in genome editing and beyond. Annu Rev Biochem 2016, 85(1): 227-264.
- Gaj T, Gersbach CA, Barbas CF: ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol 2013, 31(7): 397-405.
- Horvath P, Barrangou R: CRISPR/Cas, the immune system of bacteria and archaea. Science 2010, 327(5962): 167-170.
- Jiang F, Doudna JA: CRISPR–Cas9 structures and mechanisms. Annu Rev Biophys 2017, 46(1): 505-529.
- Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF et al: Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 2014, 371(16): 1507-1517.
- Labanieh L, Majzner RG, Mackall CL: Programming CAR-T cells to kill cancer. Nat Biomed Eng 2018, 2(6): 377-391.
- Torikai H, Cooper LJ: Translational implications for off-the-shelf immune cells expressing chimeric antigen receptors. Mol Ther 2016, 24(7): 1178-1186.
- Ruella M, Xu J, Barrett DM, Fraietta JA, Reich TJ, Ambrose DE, Klichinsky M, Shestova O, Patel PR, Kulikovskaya I et al: Induction of resistance to chimeric antigen receptor T cell therapy by transduction of a single leukemic B cell. Nat Med 2018, 24(10): 1499-1503.
- Dubrot J, Du PP, Lane-Reticker SK, Kessler EA, Muscato AJ, Mehta A, Freeman SS, Allen PM, Olander KE, Ockerman KM et al: In vivo CRISPR screens reveal the landscape of immune evasion pathways across cancer. Nat Immunol 2022, 23(10): 1495-1506.
- Li F, Huang Q, Luster TA, Hu H, Zhang H, Ng WL, Khodadadi-Jamayran A, Wang W, Chen T, Deng J et al: In vivo epigenetic CRISPR screen identifies Asf1a as an immunotherapeutic target in Kras-mutant lung adenocarcinoma. Cancer Discov 2020, 10(2): 270-287.
- Wang G, Chow RD, Zhu L, Bai Z, Ye L, Zhang F, Renauer PA, Dong MB, Dai X, Zhang X et al: CRISPR-GEMM pooled mutagenic screening identifies KMT2D as a major modulator of immune checkpoint blockade. Cancer Discov 2020, 10(12): 1912-1933.
- Kamber RA, Nishiga Y, Morton B, Banuelos AM, Barkal AA, Vences-Catalán F, Gu M, Fernandez D, Seoane JA, Yao D et al: Inter-cellular CRISPR screens reveal regulators of cancer cell phagocytosis. Nature 2021, 597(7877): 549-554.
- Chapman NM, Chi H: Metabolic adaptation of lymphocytes in immunity and disease. Immunity 2022, 55(1): 14-30.
- Fu G, Guy CS, Chapman NM, Palacios G, Wei J, Zhou P, Long L, Wang YD, Qian C, Dhungana Y et al: Metabolic control of TFH cells and humoral immunity by phosphatidylethanolamine. Nature 2021, 595(7869): 724-729.
- Sutra Del Galy A, Menegatti S, Fuentealba J, Lucibello F, Perrin L, Helft J, Darbois A, Saitakis M, Tosello J, Rookhuizen D et al: In vivo genome-wide CRISPR screens identify SOCS1 as intrinsic checkpoint of CD4+ TH1 cell response. Sci Immunol 2021, 6(66): eabe8219.
- Sugiura A, Andrejeva G, Voss K, Heintzman DR, Xu X, Madden MZ, Ye X, Beier KL, Chowdhury NU, Wolf MM et al: MTHFD2 is a metabolic checkpoint controlling effector and regulatory T cell fate and function. Immunity 2022, 55(1): 65-81.
- Ye L, Park JJ, Dong MB, Yang Q, Chow RD, Peng L, Du Y, Guo J, Dai X, Wang G et al: In vivo CRISPR screening in CD8 T cells with AAV–Sleeping Beauty hybrid vectors identifies membrane targets for improving immunotherapy for glioblastoma. Nat Biotechnol 2019, 37(11): 1302-1313.
- Marshall E: Gene therapy death prompts review of adenovirus vector. Science 1999, 286(5448): 2244-2245.
- Ewaisha R, Anderson KS: Immunogenicity of CRISPR therapeutics-Critical considerations for clinical translation. Front Bioeng Biotechnol 2023, 11: 1138596.
- Lu Y, Xue J, Deng T, Zhou X, Yu K, Deng L, Huang M, Yi X, Liang M, Wang Y et al: Safety and feasibility of CRISPR-edited T cells in patients with refractory non-small-cell lung cancer. Nat Med 2020, 26(5): 732-740.
- Stadtmauer EA, Fraietta JA, Davis MM, Cohen AD, Weber KL, Lancaster E, Mangan PA, Kulikovskaya I, Gupta M, Chen F et al: CRISPR-engineered T cells in patients with refractory cancer. Science 2020, 367(6481): eaba7365.
- Lacey SF, Fraietta JA: First trial of CRISPR-edited T cells in lung cancer. Trends Mol Med 2020, 26(8): 713-715.
- Wang Z, Li N, Feng K, Chen M, Zhang Y, Liu Y, Yang Q, Nie J, Tang N, Zhang X et al: Phase I study of CAR-T cells with PD-1 and TCR disruption in mesothelin-positive solid tumors. Cell Mol Immunol 2021, 18(9): 2188-2198.
- Zarour HM: Reversing T-cell dysfunction and exhaustion in cancer. Clin Cancer Res 2016, 22(8): 1856-1864.
- Pollari M, Brück O, Pellinen T, Vähämurto P, Karjalainen-Lindsberg ML, Mannisto S, Kallioniemi O, Kellokumpu-Lehtinen PL, Mustjoki S, Leivonen SK et al: PD-L1+ tumor-associated macrophages and PD-1+ tumor-infiltrating lymphocytes predict survival in primary testicular lymphoma. Haematologica 2018, 103(11): 1908-1914.
- Qin S, Xu L, Yi M, Yu S, Wu K, Luo S: Novel immune checkpoint targets: moving beyond PD-1 and CTLA-4. Mol Cancer 2019, 18: 1-4.
- Rowshanravan B, Halliday N, Sansom DM: CTLA-4: a moving target in immunotherapy. Blood 2018 131(1): 58-67.
- Krummel MF, Allison JP: CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med 1995, 182(2): 459-465.
- Hodi FS, O'day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC et al: Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010, 363(8): 711-723.
- Beer TM, Kwon ED, Drake CG, Fizazi K, Logothetis C, Gravis G, Ganju V, Polikoff J, Saad F, Humanski P et al: Randomized, double-blind, phase III trial of ipilimumab versus placebo in asymptomatic or minimally symptomatic patients with metastatic chemotherapy-naive castration-resistant prostate cancer. J Clin Oncol 2017, 35(1): 40-47.
- Shi L, Meng T, Zhao Z, Han J, Zhang W, Gao F, Cai J: CRISPR knock out CTLA-4 enhances the anti-tumor activity of cytotoxic T lymphocytes. Gene 2017, 636: 36-41.
- Zhang W, Shi L, Zhao Z, Du P, Ye X, Li D, Cai Z, Han J, Cai J: Disruption of CTLA-4 expression on peripheral blood CD8+ T cell enhances anti-tumor efficacy in bladder cancer. Cancer Chemother Pharmacol 2019, 83: 911-920.
- Keir ME, Butte MJ, Freeman GJ, Sharpe AH: PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol 2008, 26(1): 677-704.
- Alsaab HO, Sau S, Alzhrani R, Tatiparti K, Bhise K, Kashaw SK, Iyer AK: PD-1 and PD-L1 checkpoint signaling inhibition for cancer immunotherapy: mechanism, combinations, and clinical outcome. Front Pharmacol 2017, 8: 561.
- Sharma P, Retz M, Siefker-Radtke A, Baron A, Necchi A, Bedke J, Plimack ER, Vaena D, Grimm MO, Bracarda S et al: Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): a multicentre, single-arm, phase 2 trial. Lancet Oncol 2017, 18(3): 312-322.
- Guo X, Jiang H, Shi B, Zhou M, Zhang H, Shi Z, Du G, Luo H, Wu X, Wang Y et al: Disruption of PD-1 enhanced the anti-tumor activity of chimeric antigen receptor T cells against hepatocellular carcinoma. Front Pharmacol 2018, 9: 1118.
- Su S, Zou Z, Chen F, Ding N, Du J, Shao J, Li L, Fu Y, Hu B, Yang Y et al: CRISPR-Cas9-mediated disruption of PD-1 on human T cells for adoptive cellular therapies of EBV positive gastric cancer. Oncoimmunology 2017, 6(1): e1249558.
- Lu Y, Xue J, Deng T, Zhou X, Yu K, Deng L, Huang M, Yi X, Liang M, Wang Y et al: Safety and feasibility of CRISPR-edited T cells in patients with refractory non-small-cell lung cancer. Nat Med 2020, 26(5): 732-740.
- Tu K, Deng H, Kong L, Wang Y, Yang T, Hu Q, Hu M, Yang C, Zhang Z: Reshaping tumor immune microenvironment through acidity-responsive nanoparticles featured with CRISPR/Cas9-mediated programmed death-ligand 1 attenuation and chemotherapeutics-induced immunogenic cell death. ACS Appl Mater Interfaces 2020, 12(14): 16018-16030.
- Zhao L, Luo Y, Huang Q, Cao Z, Yang X: Photo‐enhanced CRISPR/Cas9 system enables robust pd‐l1 gene disruption in cancer cells and cancer stem‐like cells for efficient cancer immunotherapy. Small 2020, 16(52): 2004879.
- Li L, He ZY, Wei XW, Gao GP, Wei YQ: Challenges in CRISPR/CAS9 delivery: potential roles of nonviral vectors. Hum Gene Ther 2015, 26(7): 452-462.
- Nelson CE, Gersbach CA: Engineering delivery vehicles for genome editing. Annu Rev Chem Biomol Eng 2016, 7(1): 637-662.
- Chandrasekaran AP, Song M, Kim KS, Ramakrishna S: Different methods of delivering CRISPR/Cas9 into cells. Prog Mol Biol Transl Sci 2018, 159: 157-176.
- Sun W, Ji W, Hall JM, Hu Q, Wang C, Beisel CL, Gu Z: Self‐assembled DNA nanoclews for the efficient delivery of CRISPR–Cas9 for genome editing. Angew Chem Int Ed Engl 2015, 54(41): 12029-12033.
- Pineda M, Moghadam F, Ebrahimkhani MR, Kiani S: Engineered CRISPR systems for next generation gene therapies. ACS Synth Biol 2017, 6(9): 1614-1626.
- Memi F, Ntokou A, Papangeli I: CRISPR/Cas9 gene-editing: Research technologies, clinical applications and ethical considerations. Semi Perinatol 2018, 42(8): 487-500.
- Haapaniemi E, Botla S, Persson J, Schmierer B, Taipale J: CRISPR–Cas9 genome editing induces a p53-mediated DNA damage response. Nat Med 2018, 24(7): 927-930.
- Opar A: CRISPR-edited babies arrived, and regulators are still racing to catch up. Nat Med 2019, 5(11): 1634-1636.
- Petrucelli N, Daly MB, Pal T: BRCA1- and BRCA2-Associated Hereditary Breast and Ovarian Cancer. 1998 Sep 4 [Updated 2022 May 26]. In GeneReviews®; Adam, M.P., Everman, D.B., Mirzaa, G.M., Eds.; University of Washington: Seattle, WA, USA, 1993–2023.
- Guha T, Malkin D: Inherited TP53 mutations and the Li–Fraumeni syndrome. Cold Spring Harb Perspect Med 2017, 7(4): a026187.
- Chen M, Mao A, Xu M, Weng Q, Mao J, Ji J: CRISPR-Cas9 for cancer therapy: Opportunities and challenges. Cancer Lett 2019, 447: 48-55.
- Takeda H, Kataoka S, Nakayama M, Ali MA, Oshima H, Yamamoto D, Park JW, Takegami Y, An T, Jenkins NA et al: CRISPR-Cas9–mediated gene knockout in intestinal tumor organoids provides functional validation for colorectal cancer driver genes. Proc Natl Acad Sci 2019, 116(31): 15635-15644.
- Day JW, Finkel RS, Chiriboga CA, Connolly AM, Crawford TO, Darras BT, Iannaccone ST, Kuntz NL, Peña LD, Shieh PB et al: Onasemnogene abeparvovec gene therapy for symptomatic infantile-onset spinal muscular atrophy in patients with two copies of SMN2 (STR1VE): an open-label, single-arm, multicentre, phase 3 trial. Lancet Neurol 2021, 20(4): 284-293.
- Manguso RT, Pope HW, Zimmer MD, Brown FD, Yates KB, Miller BC, Collins NB, Bi K, LaFleur MW, Juneja VR et al: In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 2017 547(7664): 413-418.
- Gonzalez-Salinas F, Rojo R, Martinez-Amador C, Herrera-Gamboa J, Trevino V: Transcriptomic and cellular analyses of CRISPR/Cas9-mediated edition of FASN show inhibition of aggressive characteristics in breast cancer cells. Biochem Biophys Res Commun 2020, 529(2): 321-327.
- U.S. Food & Drug Administration. FDA Approves Novel Gene Therapy to Treat Patients with a Rare Form of Inherited Vision Loss. 2017. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-novel-gene-therapy- treat-patients-rare-form-inherited-vision-loss (accessed on 26 January 2023).
- Tang H, Shrager JB: CRISPR/Cas‐mediated genome editing to treat EGFR‐mutant lung cancer: a personalized molecular surgical therapy. EMBO Mol Med 2016, 8(2): 83-85.
- Zhang W, Shi L, Zhao Z, Du P, Ye X, Li D, Cai Z, Han J, Cai J: Disruption of CTLA-4 expression on peripheral blood CD8+ T cell enhances anti-tumor efficacy in bladder cancer. Cancer Chemother Pharmacol 2019, 83(5): 911-920.
- Russell S, Bennett J, Wellman JA, Chung DC, Yu ZF, Tillman A, Wittes J, Pappas J, Elci O, McCague S et al: Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet 2017, 390(10097): 849-860.
- Kontomanolis EN, Koutras A, Syllaios A, Schizas D, Mastoraki A, Garmpis N, Diakosavvas M, Angelou K, Tsatsaris G, Pagkalos A et al: Role of oncogenes and tumor-suppressor genes in carcinogenesis: a review. Anticancer Res 2020 40(11): 6009-6015.
- CT Coronary Angiography (CTCA): Does the DNA Editing Tool CRISPR Have a Future in Cancer Treatment? In Cancer Treatment Centers of America; 2021. Available online: https://www.cancercenter.com/community/blog/2021/04/cancer-crispr (accessed on 26 January 2023).
- Kanter J, Walters MC, Krishnamurti L, Mapara MY, Kwiatkowski JL, Rifkin-Zenenberg S, Aygun B, Kasow KA, Pierciey Jr FJ, Bonner M et al: Biologic and clinical efficacy of LentiGlobin for sickle cell disease. New Eng J Med 2022, 386(7): 617-628.
- Thompson AA, Walters MC, Kwiatkowski J, Rasko JE, Ribeil JA, Hongeng S, Magrin E, Schiller GJ, Payen E, Semeraro M et al: Gene therapy in patients with transfusion-dependent β-thalassemia. New Engl J Med 2018, 378(16): 1479-1493.
Asia-Pacific Journal of Surgical & Experimental Pathology
ISSN 2977-5817 (Online)
 Copyright © Asia Pac J Surg Exp & Pathol. This
work is licensed under a Creative Commons AttributionNonCommercial-No Derivatives 4.0 International (CC BY-NC-ND 4.0)
License.
Copyright © Asia Pac J Surg Exp & Pathol. This
work is licensed under a Creative Commons AttributionNonCommercial-No Derivatives 4.0 International (CC BY-NC-ND 4.0)
License.