
 Submit Manuscript
Submit Manuscript
Review Article | Open Access
Molecular basis of senescence in osteoarthritis
Maria G. Lawson1, Heather M. Ritchison1
1. Department of Health Sciences, Princess Nourah bint Abdulrahman University, Airport Road, Riyadh 13414, Saudi Arabia.
Correspondence: Heather M. Ritchison (Department of Health Sciences, Princess Nourah bint Abdulrahman University, Airport Road, Riyadh 13414, Saudi Arabia; E-mail: heather.ritchison@gmail.com).
Asia-Pacific Journal of Surgical & Experimental Pathology 2024, 1: 57-70. https://doi.org/10.32948/ajsep.2024.11.22
Received: 15 Aug 2024 | Accepted: 17 Nov 2024 | Published online: 26 Nov 2024
Key words osteoarthritis, senescence, DNA damage, pro-inflammatory cytokines, epigenetic modifications
Senescence, a natural aging-related response to stressors like DNA damage, telomere dysfunction, mitochondrial dysfunction, oxidative stress, autophagic stress, that introduces epigenetic, transcriptional and translational changes limiting cellular proliferation and inducing senescence associated secretory phenotype (SASP) [6, 7]. Of note, senescent joint cells express biomarkers like telomere erosion, increased expression of p53 and cyclin-dependent kinase (CDK) inhibitors p21 and p16, enhanced reactive oxygen species (ROS) generation via mitochondrial dysfunction, and senescence-associated heterochromatin, though the impact of senescence on OA is intricate and still evolving as extent and nature of senescence varies by cell types in the joint that includes the infiltrating cells from innate immune system as well [8-10]. Rather than undergoing apoptosis, these senescent cells activate pro-survival pathways like ephrins, PI3K-AKT, BCL-2 family proteins, p53-associated pathways, and FOXO4, presenting potential drug targets for OA and other senescence-related pathologies [11]. Recent findings also suggest that the establishment of cellular senescence may involve autonomous reprogramming of epigenetic mechanisms [12]. Joint tissue inflammation, a hallmark of OA, involves senescent cells contributing to the inflammatory state through the release of SASP-factors, including pro-inflammatory cytokines (interleukin-1 (IL-1), IL-8, tumor necrosis factor-alpha (TNF-α)), matrix metalloproteinases (MMPs), microRNAs (miRNAs), growth factors, and metabolites, into the tissue microenvironment [13]. Alternatively, inflammation and injury-driven immune response may also drive senescence in the affected tissue. For instance, increased Th17 T cell infiltration induces senescence through perturbation in Wnt signaling and tissue remodeling in OA [14]. Therefore, the process of senescence establishment in OA is dynamic, engaging overlapping yet distinct molecular pathways. In this review, we discuss senescence-related molecular mechanisms such as impaired DNA damage response, telomere shortening, mitochondrial dysfunction, oxidative and autophagic stresses, epigenetic modifications, and dysregulated sirtuins (SIRTs) and noncoding RNAs, contributing to the adversity of OA (Figure 1) and highlight the role of SASP as the driving force behind senescence-mediated OA progression.
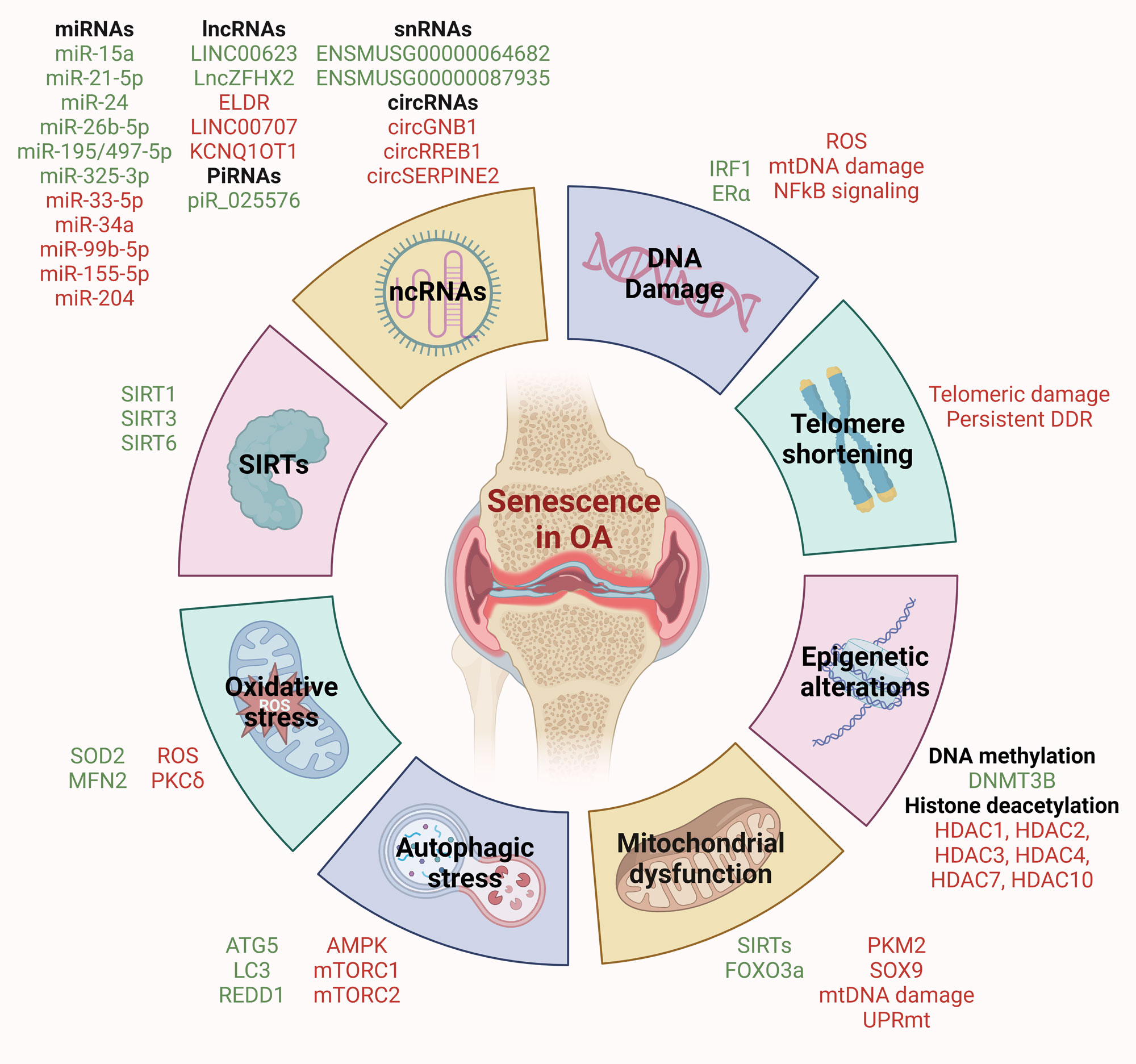 Figure 1. Molecular mechanisms driving senescence in OA. DNA damage, telomere shortening, mitochondrial dysfunction, oxidative stress, autophagic stress, epigenetic stress, and dysregulation of SIRTs and non-coding RNAs contribute to senescence in OA. Key molecular events associated with these hallmarks are listed. Red indicates upregulated/promoter factors, while green indicates downregulated/suppressor factors.
Figure 1. Molecular mechanisms driving senescence in OA. DNA damage, telomere shortening, mitochondrial dysfunction, oxidative stress, autophagic stress, epigenetic stress, and dysregulation of SIRTs and non-coding RNAs contribute to senescence in OA. Key molecular events associated with these hallmarks are listed. Red indicates upregulated/promoter factors, while green indicates downregulated/suppressor factors.
Interferon regulatory factor 1 (IRF1) plays a critical role in DNA repair in chondrocytes, and its absence leads to increased DNA damage and accelerated cellular senescence, exacerbating OA development [25]. Decline in estrogen receptor-α (ERα) levels ensues increased DNA damage and senescence markers in OA chondrocytes. Overexpression of ERα effectively reduces DNA damage and senescence in both normal and OA chondrocytes. Mechanistically, ERα overexpression partially reversed the DNA damage-induced activation of the NF-κB pathway induced by DNA damage, highlighting the critical role of ERα in maintaining chondrocyte health by inhibiting DNA damage and senescence in OA [26]. Oxidative stress induces heightened DDR that leads to senescence of chondrocytes in OA. In such scenario, knocking down IKB kinase alpha (IKKα) reduces microsatellite instability and promotes mismatch repair (MMR) proteins for better DNA damage recovery compared to what observed in IKKα deficient cells during OA development [27]. Mitochondrial DNA (mtDNA) variability serves as prognostic factor in OA, with its manipulation having potential to increase autophagy and limit oxidative stress and senescence, thereby reducing joint damage [28]. In addition, mitochondria-targeted DNA repair enzymes, like 8-oxoguanine DNA glycosylase 1 (OGG1), protect chondrocytes from mtDNA damage, promote mitochondrial transcription and maintain energy homeostasis, thereby reducing senescence and apoptosis. Pro-inflammatory cytokines often disrupt this balance by causing mitochondrial DNA damage [29]. This emphasizes the importance of maintaining mitochondrial DNA integrity, in addition to that of genomic DNA, to counteract senescence in OA.
Chondrocytes in OA patients exhibit telomere shortening and short telomere load in chondrocytes has been proposed as a significant marker for OA diagnosis and prognosis [37]. Chronic pain in OA patients also correlate with telomere length, with patients experiencing chronic severe pain showing shorter telomeres [38]. In addition, ultra-short telomere load, mean telomere length, and proximity to lesions significantly correlates to senescence level and disease severity in OA [39]. Chondrocytes from OA patients also exhibit increased chromosomal aberrations, in addition to reduced telomere length, compared to control chondrocytes, suggesting local advanced senescence in OA joints [40]. The same study also established that peripheral blood leukocytes (PBL) from OA patients exhibit increased chromosomal abnormalities, indicating a more generalized accelerated senescence phenotype associated with OA [40]. Similarly, multiple studies have also established a link between telomere shortening in PBL and OA. For instance, relative telomere length is significantly shorter in PBL from knee OA patients compared to healthy controls, and associates with high oxidative stress and inflammation [41]. In addition, individuals with knee OA at recruitment and those who developed knee OA during the follow-up have shown higher telomere loss in PBL compared to those without knee OA in a 6-year follow-up study. Telomere loss in PBL was independently associated with incident knee OA, suggesting it as a potential risk factor for accelerated cartilage degeneration in OA [42]. Furthermore, another study has correlated telomere size with age, hypertension, BMI, and waist circumference in OA patients, while affirming PBL telomere size as an independent risk factor for knee OA [43]. Meanwhile, a significant sex-by-disease-status interaction of telomere length in leukocytes, with females having greater differences than males compared to controls, has also been observed [44]. Physiological premenopausal levels of estrogen limit telomere attrition in chondrocytes, while postmenopausal estrogen levels do not do so, suggesting a potential link between estrogen decline in postmenopause and OA onset in women [45]. Severe and moderate pain in OA patients is also associated with shorter telomere length in leukocytes, highlighting sex and pain as factors influencing telomere length in OA. Moreover, cartilage telomere length in severely affected areas corresponded to disease severity, showing telomere attrition. It is noteworthy that telomeres in both severely and less affected cartilage are significantly shorter than leukocytes from the same patient [44]. Accelerated telomere loss in PBLs may indicate a systemic senescence phenotype linked to mitochondrial function, increasing the risk of developing knee OA [46]. Lastly, a meta-analysis of six studies has revealed that OA patients do present shorter telomere length in PBLs compared to healthy controls, confirming the link between telomere length in PBLs and OA pathogenesis [47]. Overall, these findings highlight the contribution of telomere shortening in driving senescence in OA.
Perturbations in proteostasis and mitochondrial metabolic functions can trigger the activation of the mitochondrial unfolded protein response (UPRmt) during stress leading to cellular senescence [55]. UPRmt activation can propagate deleterious mtDNA rearrangements, impacting oxidative phosphorylation and cellular function. Hence, controlled UPRmt activation is crucial for maintaining mitochondrial homeostasis under stress conditions [56]. At molecular level, inner mitochondrial membrane depolarization activates OMA1, leading to DELE1 cleavage. The resulting fragment activates HRI, phosphorylating eIF2α and slowing cellular protein synthesis. ATF4 and ATF5 levels increase, along with C/EBP homologous protein (CHOP), mediating the integrated stress response. SIRT1 restoration may hamper this pathway and promote chondrocyte function to alleviate osteoarthritis progression [57, 58]. SIRT1 regulates SIRT3 through PGC-1α, activating FOXO3a, that later induces SOD2 and catalase expression, reducing mitochondiral stress [59]. FOXO3a may also promote autophagy-related genes, impacting inflammatory mediators and cartilage degradation [60].
Mechanical stress-induced mitochondrial dysfunction may trigger low-grade chronic inflammation-related senescence response [61]. In turn, inflammatory factors like IL-1β and TNF-α further impair mitochondrial function, reduce energy production, and hinder mitochondrial DNA repair in chondrocytes [29, 62]. Additionally, altered redox environments in OA inhibit anabolic signaling pathways, leading to decreased cartilage matrix synthesis [63, 64]. Overall, disruptions in mitochondrial homeostasis and quality control mechanisms contribute significantly to the persistence of chondrocyte senescence and hinder recovery processes.
Oxidative stress may promote cellular senescence by instigating DNA damage and DDR [73]. In the context of OA, oxidative stress contributes significantly to disease progression by inducing genomic instability, particularly telomere instability, replicative senescence, and dysfunction in human chondrocytes [74]. For instance, hypertrophic ligamentum flavum is a characteristic of OA in spine, that is directly linked to higher levels of oxidative stress and associated telomere shortening compared to non-hypertrophic ligamentum flavum [75]. Upon exposure to oxidative stress, redox-sensitive PKCδ activates IKKα, leading to p53 phosphorylation and activation, thereby modulating DDR and influencing cell cycle arrest and senescence [76]. Despite this, the role of P53 in chondrocytes is controversial as PKR/p38 MAPK/p53/AKT/PGC-1α pathway has been shown to cause abnormal mitochondrial biosynthesis and increased oxidative stress in chondrocytes under TNF-α stimulation, wherease AMPK-SIRT-p53 signaling has been reported to protect against chondrocyte senescence [77, 78]. The latter is further supported by the findings that AMPK activation in OA chondrocytes alleviates increased mtROS associated with mtDNA4977 deletion and reduced SIRT3 expression, enhancing mitochondrial function and potentially offering chondroprotective benefits [79]. Further research is needed to clarify P53's involvement in the mitochondria of senescent chondrocytes.
During aging and OA, MFN2 expression increases in chondrocytes, promoting protective mitochondrial fusion that maintains mitochondrial function under stress [80]. However, prolonged stress disrupts mitochondrial dynamics, leading to mitochondrial dysfunction, their excessive division and compromised removal of damaged mitochondria [81]; thereby worsening chondrocyte damage induced by ROS. The interplay between inflammation and oxidative stress further exacerbates senescence-related cartilage damage in OA, with ROS playing a pivotal role in overproducing SASP components such as IL-1, IL-6, and MMPs. Inflammatory changes are associated with decreased antioxidant enzyme levels in biological fluids and cartilage, coupled with elevated oxidative agent levels, contributing to cartilage matrix protein impairment and subsequent damage [73, 82].
While the potential of targeting autophagy to modulate senescence is recognized, several complexities warrant further investigation. Autophagy's role in senescence regulation appears multifaceted, with instances where autophagy inhibition, such as through rapamycin-mediated mTOR inhibition, can prevent certain aspects of senescence like SASP induction but may not affect oncogene-induced cell cycle arrest [93]. Moreover, conflicting evidence exists regarding autophagy's overall impact on senescence, with studies suggesting both promoting and protective roles in different contexts [94]. Notably, sustained p53 activation, characteristic of senescent cells, can influence the expression of various autophagy-related proteins, indicating a potential constitutive activation of autophagy during senescence [94]. Recent studies have also linked autophagy to senescence induction via mTORC2, highlighting the intricate interplay between autophagy and senescence pathways [95]. The dual nature of autophagy in senescence underscores the need for a comprehensive understanding before considering autophagy-targeting interventions for senescence modulation in OA.
Histones are highly conserved proteins with the primary function of stabilizing, organizing, and concentrating DNA within the nucleus's limited space. They are composed of duplicate octamers that contain dimers of four core histones (H2A, H2B, H3, and H4), effectively encapsulating genomic DNA on their outer surface [99]. Acetylation plays a crucial role in modulating histone function by neutralizing the positive charge on histones, converting the amine residue into an amide. This process reduces the histone's affinity for DNA, preventing chromatin shrinkage and facilitating access for the gene transcription machinery to transcribe DNA. Conversely, deacetylation restores a positively charged histone tail, leading to high-affinity binding with the DNA backbone and resulting in chromatin condensation that blocks transcription [107]. The family of histone deacetylases (HDACs) and histone acetyltransferases are enzymes responsible for histone and non-histone deacetylation and acetylation, respectively [108]. HDAC1 and HDAC2 expression is increased in chondrocytes and synovial membranes of OA patients compared to controls. The carboxyl-terminal domain of HDAC1 and HDAC2 collaborates with the transcriptional inhibitor SNAIL to inhibit the expression of collagen α1(II) gene (COL2A1) and aggrecan, contributing to OA pathology [109, 110]. Knocking down HDAC3 in human chondrocytes results in altered expression of cartilage-related genes, potentially impacting OA progression [111]. Additionally, HDAC4, HDAC7 and HDAC10 play roles in regulating gene expression associated with cartilage degradation and inflammation, linking histone modifications to senescence in OA [112-114]. Histone methylation marks also play important role in OA pathology. For instance, knocking down Jun, whose expression is found to be reduced in senescent chondrocytes in vivo, induces micronuclei formation, reduces H3K9 trimethylation, and decreases levels of heterochromatin protein 1gamma, indicating chromatin destabilization that fosters senescence phenotype in chondrocytes [115]. Overall, epigenetic alterations play key role in acquisition of senescence in OA.
SIRT3, predominantly located in mitochondria, maintains mitochondrial homeostasis and regeneration, a phenomenon that protects against OA development [125]. In particular, SIRT3 deacetylates SOD2, a protective factor against oxidative stress, thereby increasing SOD2-specific activity and safeguarding against chondrocyte senescence and OA progression [126]. SIRT3, at the downstream of ubiquitin-specific protease 3 (USP3), also attenuates inflammation-driven chondrocyte senescence by directly deacetylating FOXO3; thereby alleviating disease progression [127]. On the other hand, SIRT6 inhibits replicative senescence and retards OA progression by reducing inflammatory responses and chondrocyte senescence [128]. Unfortunately, SIRT6 levels in chondrocytes and cartilage tissue from OA patients are substantially lower than those in normal individuals [128, 129]. This loss of SIRT6 can contribute to increased DNA damage, telomere dysfunction, and premature senescence in chondrocytes [130, 131]. Notably, IL-1β downregulates SIRT6 expression while increasing MMP13 expression, resulting in the accumulation of DNA damage and telomere dysfunction, leading to cellular senescence. SIRT6 prevents premature senescence in human chondrocytes by coordinating DNA repair mechanisms and preserving appropriate telomere function [131]. Notably, SIRT6's involvement extends to promoting DNA repair under stress and safeguarding against telomere dysfunction through its deacetylation activity [132]. In particular, SIRT6 inhibits the acetylation of p27, a protein highly acetylated with an extended half-life during cellular senescence, leading to its degradation via the ubiquitin–proteasome pathway and delaying cellular senescence [133]. In addition, overexpressing SIRT6 facilitates DNA damage repair and inhibits senescence, potentially through the activation of the Keap1/Nrf2/HO-1 signaling pathway [129].
LncRNA ELDR plays a key role in chondrocyte senescence during OA by regulating histone modifications to activate hedgehog signaling [149]. Similarly elevated LINC00707 promotes chondrocyte senescence, ECM degradation, and inflammation in OA by rescuing FSHR via sponging-off miR-330-5p [150]. LncRNA KCNQ1OT1 potentially targets and inhibit miR-1202 to promote ETS1-driven cellular senescence in OA [151]. On the other hand, LINC00623 inhibits chondrocyte apoptosis, senescence, and ECM degradation via MAPK signaling. Mechanistically, LINC00623 rescues HRAS from miR-101 mediated inhibition, resulting in hyperactive MAPK signaling and alleviating OA progression [152]. LncZFHX2 regulates RIF1 expression by forming a transcription complex with KLF4, promoting chondrocyte DNA repair and matrix homeostasis while decelerating cellular senescence [153].
Increased circGNB1 in stressed chondrocytes and aging cartilage blocks miR-152-3p, that no longer targets ring finger protein 2019 (RNF219). This stabilizes caveolin-1 (CAV1) by preventing its ubiquitination at the K47 residue, driving senescence OA progression [154]. CircRREB1 inhibits FASN degradation by impeding acetylation-mediated ubiquitination and promotes FASN stability via RanBP2-mediated SUMOylation. This CircRREB1-FASN axis suppress PI3K-AKT signaling by inhibiting FGF18/FGFR3, leading to increased p21 expression and senescence in chondrocytes [155]. Increased circSERPINE2 levels in long-term-cultured mesenchymal stem cells (MSCs) contributes to stem cell senescence. Mechanistically, circSERPINE2 interacts with YBX3, restraining it to cytoplasm. This leads to reduced PCNA transcription and, subsequent ubiquitin-mediated degradation of p21, resulting in acquisition of senescent phenotype [156]. Chondrocytes undergo replicative aging and exhibit significant changes in ncRNA expression profiles. A large scale ncRNA profiling study has identified that miR-132-5p promote chondrocyte senescence, whereas a piRNA named piR_025576, a snRNA named ENSMUSG00000064682 and a snoRNA named ENSMUSG00000087935 delay chondrocyte senescence upon overexpression [157]. Overall, different ncRNAs regulate chondrocyte senescence though varied molecular mechanisms, offering therapeutic avenues to target OA progression.
The generation of SASP factors is intricately linked to signaling pathways such as NF-κB, p38MAPK, and mTOR, operating at various levels from transcription to secretion [163]. NF-κB, a key player in inflammatory signaling, orchestrates the transcription of numerous inflammatory mediators [164] and is a pivotal pathway driving SASP factor production [165]. Cyclic GMP-AMP synthase (cGAS)-stimulator of interferon genes (STING) expression is elevated in OA tissues and chondrocytes exposed to IL-1β, promoting ECM degradation, apoptosis, and senescence. Mechanistically, STING activates the NF-κB signaling cascade, contributing to OA progression whereas its knockdown attenuates SASP production and OA development [166]. Stress-induced p38MAPK, observed in various senescent fibroblasts, plays a significant role in SASP regulation; inhibiting p38MAPK can notably reduce the secretion of most SASP factors. Moreover, p38MAPK independently induces SASP production, bypassing the DDR, thus promoting sustained SASP secretion [167]. In the context of OA, PIEZO1, and calcium ion channel component, is elevated in diseased cartilage. It increases SASP factor production, especially IL-6 and IL-1β, through activation of p38MAPK and NF-κB pathways [168]. Rapamycin, an mTOR inhibitor, modulates SASP factor production by suppressing the translation of membrane-bound IL-1α and dampening NF-κB activity [169]. Additionally, mTOR regulates the translation of MAPKAPK2 (MK2), which in turn inhibits ZFP36L1, a protein involved in degrading SASP factor transcripts, thus stabilizing SASP components [93]. Both oxidative and inflammatory stresses can induce senescence and SASP in chondrocytes with latter exhibiting a more profound impact [170]. It's important to note that the production of SASP in OA is not limited to senescent chondrocytes alone. Cells such as osteoblasts, synovial fibroblasts, synovial macrophages, and NK cells within the joints also contribute significantly to this inflammatory milieu that contributes to cartilage degradation [171]. Synovial cells, for instance, account for approximately 55% of cytokine production, and interactions between synoviocytes and chondrocytes play a critical role in OA pathogenesis [172].
SASP factors form a complex regulatory network with both upstream and downstream interactions. For instance, cell membrane-bound IL-1α enhances the DNA binding ability of NF-κB and C/EBPβ, leading to increased transcription of IL-6 and IL-8 [173]. Key SASP factors like IL-6 and IL-8 further promote SASP factor secretion, creating a positive feedback loop that reinforces senescence [174]. Lumican, an ECM glycoprotein is upregulated in OA cartilage, aggravates inflammation by enhancing proinflammatory activation via TLR4 and macrophage polarization [175]. On the other hand, ERCC1 is decreased in OA cartilage and is associated with increased expression of MMP-13 and decreased collagen type II, along with increased apoptosis and cellular senescence, highlighting ERCC1's role in protecting chondrocytes from SASP-driven disease progression [176].
Early SASP factors induce senescence in neighboring normal cells, thereby contributing to senescence expansion, whereas later SASP factors recruit immune cells and intensify the inflammatory milieu [177]. In the context of OA, EVs from human OA-derived chondrocytes, enriched with Cx43, have been shown to induce a senescent phenotype and activate inflammatory pathways in targeted cells, promoting joint degeneration through SASP. These EVs alter protein profiles and cellular plasticity, and activate pro-survival and pro-inflammatory mechanisms such as ERK1/2 and NF-κB signaling pathways, highlighting their role in disease progression by spreading senescence, inflammation, and reprogramming factors across cartilage, synovium, and bone, potentially affecting multiple joints [178]. Senescent microenvironment also impedes cartilage repair by bone marrow stem cells (BMSCs). Although BMSCs have potential to induce apoptosis in senescent chondrocytes and reduce their numbers, senescent chondrocytes inhibit BMSC proliferation, promote senescence, and suppress chondrogenic differentiation; thereby inhibiting cartilage repair by BMSCs [179]. In summary, SASP drives a complex network of inflammatory mediators, ECM remodeling enzymes, and signaling pathways, contributing to the reinforcement of senescence through systemic effects. Understanding these intricate mechanisms is crucial for developing targeted interventions to modulate the SASP-driven inflammatory milieu and promote tissue repair in OA.
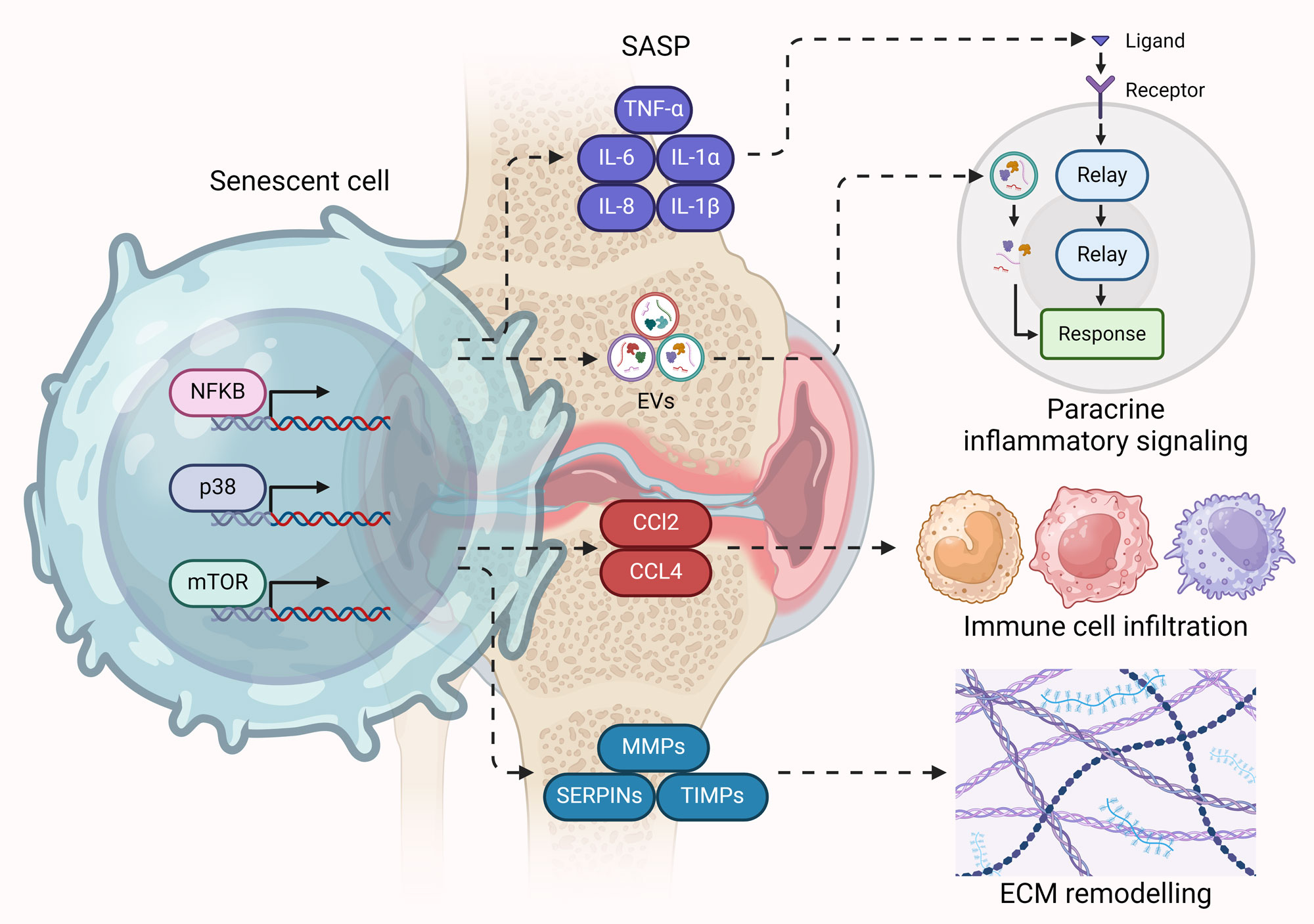 Figure 2. SASP – The driving force behind senescence-mediated OA progression. Hyperactive NF-κB, p38, and mTOR signaling in senescent cells (particularly chondrocytes) in OA drive the expression of pro-inflammatory cytokines, chemoattractants, ECM remodelers, and aberrant release of EVs as part of SASP. Pro-inflammatory cytokines and EVs promote inflammation and senescence in neighboring cells through paracrine signaling. Chemoattractants promote immune cell infiltration in the cartilage, while ECM remodelers disrupt ECM homeostasis.
Figure 2. SASP – The driving force behind senescence-mediated OA progression. Hyperactive NF-κB, p38, and mTOR signaling in senescent cells (particularly chondrocytes) in OA drive the expression of pro-inflammatory cytokines, chemoattractants, ECM remodelers, and aberrant release of EVs as part of SASP. Pro-inflammatory cytokines and EVs promote inflammation and senescence in neighboring cells through paracrine signaling. Chemoattractants promote immune cell infiltration in the cartilage, while ECM remodelers disrupt ECM homeostasis.
No applicable.
Ethics approval
No applicable.
Data availability
The data will be available upon request.
Funding
None.
Authors’ contribution
Maria G. Lawson contributed to the conception, design, writing of this review article and submitted the final version of the manuscript. Heather M. Ritchison work on the figure production and figure legends.
Competing interests
None.
- Dillon CF, Rasch EK, Gu Q, Hirsch R: Prevalence of knee osteoarthritis in the United States: arthritis data from the Third National Health and Nutrition Examination Survey 1991-94. J Rheumatol 2006, 33(11): 2271-2279.
- Buckwalter JA, Saltzman C, Brown T: The impact of osteoarthritis: implications for research. Clin Orthop Relat Res 2004, 427: S6-15.
- Honkonen SE: Degenerative arthritis after tibial plateau fractures. J Orthop Trauma 1995, 9(4): 273-277.
- Lotz M, Loeser RF: Effects of aging on articular cartilage homeostasis. Bone 2012, 51(2): 241-248.
- Martin JA, Buckwalter JA: The role of chondrocyte senescence in the pathogenesis of osteoarthritis and in limiting cartilage repair. J Bone Joint Surg Am 2003, 85-A Suppl 2: 106-110.
- Di Micco R, Krizhanovsky V: Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat Rev Mol Cell Biol 2021, 22(2): 75-95.
- Coppé JP, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J: Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 2008, 6(12): 2853-2868.
- Diekman BO, Sessions GA, Collins JA, Knecht AK, Strum SL, Mitin NK, Carlson CS, Loeser RF, Sharpless NE: Expression of p16 INK 4a is a biomarker of chondrocyte aging but does not cause osteoarthritis. Aging cell 2018, 17(4): e12771.
- Shimada H, Sakakima H, Tsuchimochi K, Matsuda F, Komiya S, Goldring MB, Ijiri K: Senescence of chondrocytes in aging articular cartilage: GADD45β mediates p21 expression in association with C/EBPβ in senescence-accelerated mice. Pathol Res Pract 2011, 207(4): 225-231.
- Loeser RF: Aging and osteoarthritis: the role of chondrocyte senescence and aging changes in the cartilage matrix. Osteoarthritis Cartilage 2009, 17(8): 971-979.
- Astrike-Davis EM, Coryell P, Loeser RF: Targeting cellular senescence as a novel treatment for osteoarthritis. Curr Opin Pharmacol 2022, 64: 102213.
- Milanovic M, Fan DN, Belenki D, Däbritz JHM, Zhao Z, Yu Y, Dörr JR, Dimitrova L, Lenze D, Monteiro Barbosa IA: Senescence-associated reprogramming promotes cancer stemness. Nature 2018, 553(7686): 96-100.
- Gasek NS, Kuchel GA, Kirkland JL, Xu M: Strategies for Targeting Senescent Cells in Human Disease. Nat Aging 2021, 1(10): 870-879.
- Faust HJ, Zhang H, Han J, Wolf MT, Jeon OH, Sadtler K, Peña AN, Chung L, Maestas DR, Jr., Tam AJ et al: IL-17 and immunologically induced senescence regulate response to injury in osteoarthritis. J Clin Invest 2020, 130(10): 5493-5507.
- Ovadya Y, Landsberger T, Leins H, Vadai E, Gal H, Biran A, Yosef R, Sagiv A, Agrawal A, Shapira A: Impaired immune surveillance accelerates accumulation of senescent cells and aging. Nat Commun 2018, 9(1): 5435.
- Jackson SP, Bartek J: The DNA-damage response in human biology and disease. Nature 2009, 461(7267): 1071-1078.
- Fumagalli M, Rossiello F, Mondello C, d’Adda di Fagagna F: Stable cellular senescence is associated with persistent DDR activation. PloS one 2014, 9(10): e110969.
- Galbiati A, Beauséjour C, d'Adda di Fagagna F: A novel single‐cell method provides direct evidence of persistent DNA damage in senescent cells and aged mammalian tissues. Aging cell 2017, 16(2): 422-427.
- Beauséjour CM, Krtolica A, Galimi F, Narita M, Lowe SW, Yaswen P, Campisi J: Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J 2003, 22(16): 4212-4222.
- Dulić V, Beney GE, Frebourg G, Drullinger LF, Stein GH: Uncoupling between phenotypic senescence and cell cycle arrest in aging p21-deficient fibroblasts. Mol Cell Biol 2000, 20(18): 6741-6754.
- Kamijo T, Zindy F, Roussel MF, Quelle DE, Downing JR, Ashmun RA, Grosveld G, Sherr CJ: Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell 1997, 91(5): 649-659.
- Fagagna FdAd, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, Saretzki G, Carter NP, Jackson SP: A DNA damage checkpoint response in telomere-initiated senescence. Nature 2003, 426(6963): 194-198.
- Mallette FA, Ferbeyre G: The DNA damage signaling pathway connects oncogenic stress to cellular senescence. Cell cycle 2007, 6(15): 1831-1836.
- Rose J, Söder S, Skhirtladze C, Schmitz N, Gebhard PM, Sesselmann S, Aigner T: DNA damage, discoordinated gene expression and cellular senescence in osteoarthritic chondrocytes. Osteoarthritis Cartilage 2012, 20(9): 1020-1028.
- Cho Y, Kim H: Predisposal of Interferon Regulatory Factor 1 Deficiency to Accumulate DNA Damage and Promote Osteoarthritis Development in Cartilage. Arthritis Rheumatol 2024, 76(6): 882-893.
- Zhang X, Xiang S, Zhang Y, Liu S, Lei G, Hines S, Wang N, Lin H: In vitro study to identify ligand-independent function of estrogen receptor-α in suppressing DNA damage-induced chondrocyte senescence. FASEB J 2023, 37(2): e22746.
- Neri S, Guidotti S, Bini C, Pelotti S, D'Adamo S, Minguzzi M, Platano D, Santi S, Mariani E, Cattini L et al: Oxidative stress-induced DNA damage and repair in primary human osteoarthritis chondrocytes: focus on IKKα and the DNA Mismatch Repair System. Free Radic Biol Med 2021, 166: 212-225.
- Scotece M, Vaamonde-García C, Lechuga-Vieco AV, Cortés AC, Gómez MCJ, Filgueira-Fernández P, Rego-Pérez I, Enríquez JA, Blanco FJ: mtDNA variability determines spontaneous joint aging damage in a conplastic mouse model. Aging (Albany NY) 2022, 14(15): 5966-5983.
- Kim J, Xu M, Xo R, Mates A, Wilson GL, Pearsall AWt, Grishko V: Mitochondrial DNA damage is involved in apoptosis caused by pro-inflammatory cytokines in human OA chondrocytes. Osteoarthritis Cartilage 2010, 18(3): 424-432.
- d'Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, Saretzki G, Carter NP, Jackson SP: A DNA damage checkpoint response in telomere-initiated senescence. Nature 2003, 426(6963): 194-198.
- Herbig U, Jobling WA, Chen BP, Chen DJ, Sedivy JM: Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a). Mol Cell 2004, 14(4): 501-513.
- Hemann MT, Strong MA, Hao LY, Greider CW: The shortest telomere, not average telomere length, is critical for cell viability and chromosome stability. Cell 2001, 107(1): 67-77.
- Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, Harley CB, Shay JW, Lichtsteiner S, Wright WE: Extension of life-span by introduction of telomerase into normal human cells. Science 1998, 279(5349): 349-352.
- Fumagalli M, Rossiello F, Clerici M, Barozzi S, Cittaro D, Kaplunov JM, Bucci G, Dobreva M, Matti V, Beausejour CM et al: Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat Cell Biol 2012, 14(4): 355-365.
- Hewitt G, Jurk D, Marques FD, Correia-Melo C, Hardy T, Gackowska A, Anderson R, Taschuk M, Mann J, Passos JF: Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat Commun 2012, 3: 708.
- Rossiello F, Aguado J, Sepe S, Iannelli F, Nguyen Q, Pitchiaya S, Carninci P: DNA damage response inhibition at dysfunctional telomeres by modulation of telomeric DNA damage response RNAs. Nat Commun 2017, 8: 13980.
- Harbo M, Delaisse JM, Kjaersgaard-Andersen P, Soerensen FB, Koelvraa S, Bendix L: The relationship between ultra-short telomeres, aging of articular cartilage and the development of human hip osteoarthritis. Mech Ageing Dev 2013, 134(9): 367-372.
- Sibille KT, Chen H, Bartley EJ, Riley J, 3rd, Glover TL, King CD, Zhang H, Cruz-Almeida Y, Goodin BR, Sotolongo A et al: Accelerated aging in adults with knee osteoarthritis pain: consideration for frequency, intensity, time, and total pain sites. Pain Rep 2017, 2(3): e591.
- Harbo M, Bendix L, Bay-Jensen AC, Graakjaer J, Søe K, Andersen TL, Kjaersgaard-Andersen P, Koelvraa S, Delaisse JM: The distribution pattern of critically short telomeres in human osteoarthritic knees. Arthritis Res Ther 2012, 14(1): R12.
- Tamayo M, Mosquera A, Rego I, Blanco FJ, Gosálvez J, Fernández JL: Decreased length of telomeric DNA sequences and increased numerical chromosome aberrations in human osteoarthritic chondrocytes. Mutat Res 2011, 708(1-2): 50-58.
- Poonpet T, Saetan N, Tanavalee A, Wilairatana V, Yuktanandana P, Honsawek S: Association between leukocyte telomere length and angiogenic cytokines in knee osteoarthritis. Int J Rheum Dis 2018, 21(1): 118-125.
- Guillén R, Otero F, Mosquera A, Vázquez-Mosquera M, Rego-Pérez I, Blanco FJ, Fernández JL: Association of accelerated dynamics of telomere sequence loss in peripheral blood leukocytes with incident knee osteoarthritis in Osteoarthritis Initiative cohort. Sci Rep 2021, 11(1): 15914.
- Mosquera A, Rego-Pérez I, Blanco FJ, Fernández JL: Leukocyte Telomere Length in Patients with Radiographic Knee Osteoarthritis. Environ Mol Mutagen 2019, 60(3): 298-301.
- Kuszel L, Trzeciak T, Begier-Krasinska B, Richter M, Li J, Czarny-Ratajczak M: Sex-specific differences in telomere length of patients with primary knee osteoarthritis. J Cell Mol Med 2024, 28(3): e18107.
- Breu A, Sprinzing B, Merkl K, Bechmann V, Kujat R, Jenei-Lanzl Z, Prantl L, Angele P: Estrogen reduces cellular aging in human mesenchymal stem cells and chondrocytes. J Orthop Res 2011, 29(10): 1563-1571.
- Fajardo RG, Fariña FO: Relationship Between the Dynamics of Telomere Loss in Peripheral Blood Leukocytes From Knee Osteoarthritis Patients and Mitochondrial DNA Haplogroups. J Rheumatol 2021, 48(10): 1603-1607.
- Xie H, Ma Y, Shao M, Kong J, Zhou T, Wang F, Cai G, Xu S, Pan F: Telomere length in patients with osteoarthritis: a systematic review and meta-analysis. Aging Clin Exp Res 2022, 34(3): 495-503.
- Chapman J, Fielder E, Passos JF: Mitochondrial dysfunction and cell senescence: deciphering a complex relationship. FEBS Lett 2019, 593(13): 1566-1579.
- Wiley CD, Velarde MC, Lecot P, Liu S, Sarnoski EA, Freund A, Shirakawa K, Lim HW, Davis SS, Ramanathan A et al: Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab 2016, 23(2): 303-314.
- Correia-Melo C, Marques FD, Anderson R, Hewitt G, Hewitt R, Cole J, Carroll BM, Miwa S, Birch J, Merz A et al: Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J 2016, 35(7): 724-742.
- Blanco FJ, Rego I, Ruiz-Romero C: The role of mitochondria in osteoarthritis. Nat Rev Rheumatol 2011, 7(3): 161-169.
- Ashraf S, Cha BH, Kim JS, Ahn J, Han I, Park H, Lee SH: Regulation of senescence associated signaling mechanisms in chondrocytes for cartilage tissue regeneration. Osteoarthritis Cartilage 2016, 24(2): 196-205.
- Shapiro IM, Srinivas V: Metabolic consideration of epiphyseal growth: survival responses in a taxing environment. Bone 2007, 40(3): 561-567.
- Yang X, Chen W, Zhao X, Chen L, Li W, Ran J, Wu L: Pyruvate Kinase M2 Modulates the Glycolysis of Chondrocyte and Extracellular Matrix in Osteoarthritis. DNA Cell Biol 2018, 37(3): 271-277.
- Baumann K: Ageing: The yin and yang of mitochondrial dysfunction. Nat Rev Mol Cell Biol 2016, 17(6): 331.
- Lin YF, Schulz AM, Pellegrino MW, Lu Y, Shaham S, Haynes CM: Maintenance and propagation of a deleterious mitochondrial genome by the mitochondrial unfolded protein response. Nature 2016, 533(7603): 416-419.
- Feng K, Ge Y: Curcumin Inhibits the PERK-eIF2α-CHOP Pathway through Promoting SIRT1 Expression in Oxidative Stress-induced Rat Chondrocytes and Ameliorates Osteoarthritis Progression in a Rat Model. Oxid Med Cell Longev 2019, 2019: 8574386.
- Lin Z, Teng C, Ni L, Zhang Z, Lu X, Lou J, Wang L, Wang Y, Chen W, Zhang X: Echinacoside Upregulates Sirt1 to Suppress Endoplasmic Reticulum Stress and Inhibit Extracellular Matrix Degradation In Vitro and Ameliorates Osteoarthritis In Vivo. Oxid Med Cell Longev 2021, 2021: 3137066.
- Zhang Y, Dai J, Yan L, Lin Q, Miao H, Wang X, Wang J: DL-3-N-Butylphthalide Promotes Cartilage Extracellular Matrix Synthesis and Inhibits Osteoarthritis Development by Regulating FoxO3a. Oxid Med Cell Longev 2022, 2022: 9468040.
- Matsuzaki T, Alvarez-Garcia O: FoxO transcription factors modulate autophagy and proteoglycan 4 in cartilage homeostasis and osteoarthritis. Sci Transl Med 2018, 10(428): eaan0746.
- Ziegler DV, Wiley CD, Velarde MC: Mitochondrial effectors of cellular senescence: beyond the free radical theory of aging. Aging Cell 2015, 14(1): 1-7.
- Grishko VI, Ho R, Wilson GL, Pearsall AWt: Diminished mitochondrial DNA integrity and repair capacity in OA chondrocytes. Osteoarthritis Cartilage 2009, 17(1): 107-113.
- Bolduc JA, Collins JA, Loeser RF: Reactive oxygen species, aging and articular cartilage homeostasis. Free Radic Biol Med 2019, 132: 73-82.
- Loeser RF, Gandhi U, Long DL, Yin W, Chubinskaya S: Aging and oxidative stress reduce the response of human articular chondrocytes to insulin-like growth factor 1 and osteogenic protein 1. Arthritis Rheumatol 2014, 66(8): 2201-2209.
- Loeser RF, Collins JA, Diekman BO: Ageing and the pathogenesis of osteoarthritis. Nat Rev Rheumatol 2016, 12(7): 412-420.
- Passos JF, Saretzki G, von Zglinicki T: DNA damage in telomeres and mitochondria during cellular senescence: is there a connection? Nucleic Acids Res 2007, 35(22): 7505-7513.
- Almeida M, O'Brien CA: Basic biology of skeletal aging: role of stress response pathways. J Gerontol A Biol Sci Med Sci 2013, 68(10): 1197-1208.
- Frenkel SR, Clancy RM, Ricci JL, Di Cesare PE, Rediske JJ, Abramson SB: Effects of nitric oxide on chondrocyte migration, adhesion, and cytoskeletal assembly. Arthritis Rheum 1996, 39(11): 1905-1912.
- Johnson K, Jung A, Murphy A, Andreyev A, Dykens J, Terkeltaub R: Mitochondrial oxidative phosphorylation is a downstream regulator of nitric oxide effects on chondrocyte matrix synthesis and mineralization. Arthritis Rheum 2000, 43(7): 1560-1570.
- Koike M, Nojiri H, Ozawa Y, Watanabe K, Muramatsu Y, Kaneko H, Morikawa D, Kobayashi K, Saita Y, Sasho T et al: Mechanical overloading causes mitochondrial superoxide and SOD2 imbalance in chondrocytes resulting in cartilage degeneration. Sci Rep 2015, 5: 11722.
- Zhang Y, Unnikrishnan A, Deepa SS, Liu Y, Li Y, Ikeno Y, Sosnowska D, Van Remmen H, Richardson A: A new role for oxidative stress in aging: The accelerated aging phenotype in Sod1(-/)(-) mice is correlated to increased cellular senescence. Redox Biol 2017, 11: 30-37.
- Collins JA, Wood ST, Nelson KJ, Rowe MA, Carlson CS, Chubinskaya S, Poole LB, Furdui CM, Loeser RF: Oxidative Stress Promotes Peroxiredoxin Hyperoxidation and Attenuates Pro-survival Signaling in Aging Chondrocytes. J Biol Chem 2016, 291(13): 6641-6654.
- Liu Y, Zhang Z, Li T, Xu H, Zhang H: Senescence in osteoarthritis: from mechanism to potential treatment. Arthritis Res Ther 2022, 24(1): 174.
- Yudoh K, Nguyen v T, Nakamura H, Hongo-Masuko K, Kato T, Nishioka K: Potential involvement of oxidative stress in cartilage senescence and development of osteoarthritis: oxidative stress induces chondrocyte telomere instability and downregulation of chondrocyte function. Arthritis Res Ther 2005, 7(2): R380-391.
- Dechsupa S, Yingsakmongkol W, Limthongkul W, Singhatanadgige W, Honsawek S: Relative telomere length and oxidative DNA damage in hypertrophic ligamentum flavum of lumbar spinal stenosis. PeerJ 2018, 6: e5381.
- Yamaguchi T, Miki Y, Yoshida K: Protein kinase C delta activates IkappaB-kinase alpha to induce the p53 tumor suppressor in response to oxidative stress. Cell Signal 2007, 19(10): 2088-2097.
- Bi J, Cai W, Ma T, Deng A, Ma P, Han Y, Lou C, Wu L: Protective effect of vildagliptin on TNF-α-induced chondrocyte senescence. IUBMB Life 2019, 71(7): 978-985.
- Ma CH, Wu CH, Jou IM, Tu YK, Hung CH, Chou WC, Chang YC, Hsieh PL, Tsai KL: PKR Promotes Oxidative Stress and Apoptosis of Human Articular Chondrocytes by Causing Mitochondrial Dysfunction through p38 MAPK Activation-PKR Activation Causes Apoptosis in Human Chondrocytes. Antioxidants (Basel) 2019, 8(9): 370.
- Chen LY, Wang Y, Terkeltaub R, Liu-Bryan R: Activation of AMPK-SIRT3 signaling is chondroprotective by preserving mitochondrial DNA integrity and function. Osteoarthritis Cartilage 2018, 26(11): 1539-1550.
- Xu L, Wu Z, He Y, Chen Z, Xu K, Yu W, Fang W, Ma C, Moqbel SAA, Ran J et al: MFN2 contributes to metabolic disorders and inflammation in the aging of rat chondrocytes and osteoarthritis. Osteoarthritis Cartilage 2020, 28(8): 1079-1091.
- Zhao T, Niu D, Chen Y, Fu P: The role of mitochondrial quality control mechanisms in chondrocyte senescence. Exp Gerontol 2024, 188: 112379.
- Lepetsos P, Papavassiliou AG: ROS/oxidative stress signaling in osteoarthritis. Biochim Biophys Acta 2016, 1862(4): 576-591.
- Aman Y, Schmauck-Medina T: Autophagy in healthy aging and disease. Nat Aging 2021, 1(8): 634-650.
- Caramés B, Taniguchi N, Otsuki S, Blanco FJ, Lotz M: Autophagy is a protective mechanism in normal cartilage, and its aging-related loss is linked with cell death and osteoarthritis. Arthritis Rheum 2010, 62(3): 791-801.
- López de Figueroa P, Lotz MK, Blanco FJ, Caramés B: Autophagy activation and protection from mitochondrial dysfunction in human chondrocytes. Arthritis Rheumatol 2015, 67(4): 966-976.
- Caramés B, Olmer M, Kiosses WB, Lotz MK: The relationship of autophagy defects to cartilage damage during joint aging in a mouse model. Arthritis Rheumatol 2015, 67(6): 1568-1576.
- Alvarez-Garcia O, Matsuzaki T, Olmer M, Plate L, Kelly JW, Lotz MK: Regulated in Development and DNA Damage Response 1 Deficiency Impairs Autophagy and Mitochondrial Biogenesis in Articular Cartilage and Increases the Severity of Experimental Osteoarthritis. Arthritis Rheumatol 2017, 69(7): 1418-1428.
- Moreno-Blas D, Gorostieta-Salas E, Castro-Obregón S: Connecting chaperone-mediated autophagy dysfunction to cellular senescence. Ageing Res Rev 2018, 41: 34-41.
- Kang C, Xu Q, Martin TD, Li MZ, Demaria M, Aron L, Lu T, Yankner BA, Campisi J, Elledge SJ: The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 2015, 349(6255): aaa5612.
- Terkeltaub R, Yang B, Lotz M, Liu-Bryan R: Chondrocyte AMP-activated protein kinase activity suppresses matrix degradation responses to proinflammatory cytokines interleukin-1β and tumor necrosis factor α. Arthritis Rheum 2011, 63(7): 1928-1937.
- Ansari MY, Ahmad N, Haqqi TM: Butein Activates Autophagy Through AMPK/TSC2/ULK1/mTOR Pathway to Inhibit IL-6 Expression in IL-1β Stimulated Human Chondrocytes. Cell Physiol Biochem 2018, 49(3): 932-946.
- Carames B, De Figueroa PL, Lotz M, Blanco FJ: Autophagy activation protects from mitochondrial dysfunction in human chondrocytes. Osteoarthritis Cartilage 2014, 22: S135.
- Herranz N, Gallage S, Mellone M, Wuestefeld T, Klotz S, Hanley CJ, Raguz S, Acosta JC, Innes AJ, Banito A et al: mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat Cell Biol 2015, 17(9): 1205-1217.
- Kang C, Elledge SJ: How autophagy both activates and inhibits cellular senescence. Autophagy 2016, 12(5): 898-899.
- Bernard M, Yang B, Migneault F: Autophagy drives fibroblast senescence through MTORC2 regulation. Autophagy 2020, 16(11): 2004-2016.
- Vidal-Bralo L, Lopez-Golan Y, Mera-Varela A, Rego-Perez I, Horvath S, Zhang Y, Del Real Á, Zhai G, Blanco FJ, Riancho JA et al: Specific premature epigenetic aging of cartilage in osteoarthritis. Aging (Albany NY) 2016, 8(9): 2222-2231.
- Oh ES, Petronis A: Origins of human disease: the chrono-epigenetic perspective. Nat Rev Genet 2021, 22(8): 533-546.
- Luo C, Hajkova P: Dynamic DNA methylation: In the right place at the right time. Science 2018, 361(6409): 1336-1340.
- Simon TC, Jeffries MA: The Epigenomic Landscape in Osteoarthritis. Curr Rheumatol Rep 2017, 19(6): 30.
- Barter MJ, Bui C, Cheung K, Falk J, Gómez R: DNA hypomethylation during MSC chondrogenesis occurs predominantly at enhancer regions. Sci Rep 2020, 10(1): 1169.
- Takahashi A, de Andrés MC, Hashimoto K, Itoi E, Oreffo RO: Epigenetic regulation of interleukin-8, an inflammatory chemokine, in osteoarthritis. Osteoarthritis Cartilage 2015, 23(11): 1946-1954.
- Shen J, Wang C, Li D, Xu T, Myers J, Ashton JM, Wang T, Zuscik MJ, McAlinden A, O'Keefe RJ: DNA methyltransferase 3b regulates articular cartilage homeostasis by altering metabolism. JCI Insight 2017, 2(12): e93612.
- Hashimoto K, Oreffo RO, Gibson MB, Goldring MB, Roach HI: DNA demethylation at specific CpG sites in the IL1B promoter in response to inflammatory cytokines in human articular chondrocytes. Arthritis Rheum 2009, 60(11): 3303-3313.
- Roach HI, Yamada N, Cheung KS, Tilley S, Clarke NM, Oreffo RO, Kokubun S, Bronner F: Association between the abnormal expression of matrix-degrading enzymes by human osteoarthritic chondrocytes and demethylation of specific CpG sites in the promoter regions. Arthritis Rheum 2005, 52(10): 3110-3124.
- Papageorgiou AA, Litsaki M, Mourmoura E, Papathanasiou I, Tsezou A: DNA methylation regulates Sirtuin 1 expression in osteoarthritic chondrocytes. Adv Med Sci 2023, 68(1): 101-110.
- Liu W, Brodsky AS, Feng M, Liu Y, Ding J, Jayasuriya CT, Chen Q: Senescent Tissue-Resident Mesenchymal Stromal Cells Are an Internal Source of Inflammation in Human Osteoarthritic Cartilage. Front Cell Dev Biol 2021, 9: 725071.
- Wan C, Zhang F, Yao H, Li H, Tuan RS: Histone Modifications and Chondrocyte Fate: Regulation and Therapeutic Implications. Front Cell Dev Biol 2021, 9: 626708.
- Millán-Zambrano G, Burton A, Bannister AJ, Schneider R: Histone post-translational modifications - cause and consequence of genome function. Nat Rev Genet 2022, 23(9): 563-580.
- Huber LC, Brock M, Hemmatazad H, Giger OT, Moritz F, Trenkmann M, Distler JH, Gay RE, Kolling C, Moch H et al: Histone deacetylase/acetylase activity in total synovial tissue derived from rheumatoid arthritis and osteoarthritis patients. Arthritis Rheum 2007, 56(4): 1087-1093.
- Hong S, Derfoul A, Pereira-Mouries L, Hall DJ: A novel domain in histone deacetylase 1 and 2 mediates repression of cartilage-specific genes in human chondrocytes. FASEB J 2009, 23(10): 3539-3552.
- Meng F, Li Z, Zhang Z, Yang Z, Kang Y, Zhao X, Long D, Hu S, Gu M, He S et al: MicroRNA-193b-3p regulates chondrogenesis and chondrocyte metabolism by targeting HDAC3. Theranostics 2018, 8(10): 2862-2883.
- Higashiyama R, Miyaki S, Yamashita S, Yoshitaka T, Lindman G, Ito Y, Sasho T, Takahashi K, Lotz M, Asahara H: Correlation between MMP-13 and HDAC7 expression in human knee osteoarthritis. Mod Rheumatol 2010, 20(1): 11-17.
- Cao K, Wei L, Zhang Z, Guo L, Zhang C, Li Y, Sun C, Sun X, Wang S, Li P et al: Decreased histone deacetylase 4 is associated with human osteoarthritis cartilage degeneration by releasing histone deacetylase 4 inhibition of runt-related transcription factor-2 and increasing osteoarthritis-related genes: a novel mechanism of human osteoarthritis cartilage degeneration. Arthritis Res Ther 2014, 16(6): 491.
- Liao W, Sun J, Liu W, Li W, Jia J, Ou F, Su K, Zheng Y, Zhang Z, Sun Y: HDAC10 upregulation contributes to interleukin 1β-mediated inflammatory activation of synovium-derived mesenchymal stem cells in temporomandibular joint. J Cell Physiol 2019, 234(8): 12646-12662.
- Xie T, Ren X, Zhuang H, Jiang F, Zhang Y, Zhou P: Down-regulation of Jun induces senescence through destabilizing chromatin in osteoarthritis chondrocytes. Am J Transl Res 2023, 15(7): 4873-4886.
- Morris BJ: Seven sirtuins for seven deadly diseases of aging. Free Radic Biol Med 2013, 56: 133-171.
- Wu QJ, Zhang TN, Chen HH, Yu XF, Lv JL, Liu YY, Liu YS, Zheng G, Zhao JQ, Wei YF et al: The sirtuin family in health and disease. Signal Transduct Target Ther 2022, 7(1): 402.
- Imai S, Armstrong CM, Kaeberlein M, Guarente L: Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 2000, 403(6771): 795-800.
- Raza U, Tang X, Liu Z, Liu B: SIRT7: the seventh key to unlocking the mystery of aging. Physiol Rev 2024, 104(1): 253-280.
- Coryell PR, Diekman BO, Loeser RF: Mechanisms and therapeutic implications of cellular senescence in osteoarthritis. Nat Rev Rheumatol 2021, 17(1): 47-57.
- Dvir-Ginzberg M, Gagarina V, Lee EJ, Hall DJ: Regulation of cartilage-specific gene expression in human chondrocytes by SirT1 and nicotinamide phosphoribosyltransferase. J Biol Chem 2008, 283(52): 36300-36310.
- Xu M, Feng M, Peng H, Qian Z, Zhao L, Wu S: Epigenetic regulation of chondrocyte hypertrophy and apoptosis through Sirt1/P53/P21 pathway in surgery-induced osteoarthritis. Biochem Biophys Res Commun 2020, 528(1): 179-185.
- Batshon G, Elayyan J, Qiq O, Reich E, Ben-Aderet L, Kandel L, Haze A, Steinmeyer J, Lefebvre V, Zhang H et al: Serum NT/CT SIRT1 ratio reflects early osteoarthritis and chondrosenescence. Ann Rheum Dis 2020, 79(10): 1370-1380.
- Dudek M, Gossan N, Yang N, Im HJ, Ruckshanthi JP, Yoshitane H, Li X, Jin D, Wang P, Boudiffa M et al: The chondrocyte clock gene Bmal1 controls cartilage homeostasis and integrity. J Clin Invest 2016, 126(1): 365-376.
- He Y, Wu Z, Xu L, Xu K, Chen Z, Ran J, Wu L: The role of SIRT3-mediated mitochondrial homeostasis in osteoarthritis. Cell Mol Life Sci 2020, 77(19): 3729-3743.
- Fu Y, Kinter M, Hudson J, Humphries KM, Lane RS, White JR, Hakim M, Pan Y, Verdin E, Griffin TM: Aging Promotes Sirtuin 3-Dependent Cartilage Superoxide Dismutase 2 Acetylation and Osteoarthritis. Arthritis Rheumatol 2016, 68(8): 1887-1898.
- Zhou Q, Wang W, Wu J, Qiu S, Yuan S, Fu PL, Qian QR, Xu YZ: Ubiquitin-specific protease 3 attenuates interleukin-1β-mediated chondrocyte senescence by deacetylating forkhead box O-3 via sirtuin-3. Bioengineered 2022, 13(2): 2017-2027.
- Wu Y, Chen L, Wang Y, Li W, Lin Y, Yu D, Zhang L, Li F, Pan Z: Overexpression of Sirtuin 6 suppresses cellular senescence and NF-κB mediated inflammatory responses in osteoarthritis development. Sci Rep 2015, 5: 17602.
- Mao LW, Jiang QY, Meng N, Xiao L, Zhang Q, Chen YX, Liu LJ, Wang L: Sirt6 promotes DNA damage repair in osteoarthritis chondrocytes by activating the Keap1/Nrf2/HO-1 signaling pathway. Cell Cycle 2024, 23(2): 205-217..
- Duarte JH: Osteoarthritis: SIRT6 prevents chondrocyte senescence and DNA damage. Nat Rev Rheumatol 2015, 11(5): 260.
- Nagai K, Matsushita T, Matsuzaki T, Takayama K, Matsumoto T, Kuroda R, Kurosaka M: Depletion of SIRT6 causes cellular senescence, DNA damage, and telomere dysfunction in human chondrocytes. Osteoarthritis Cartilage 2015, 23(8): 1412-1420.
- Kawahara TL, Michishita E, Adler AS, Damian M, Berber E, Lin M, McCord RA, Ongaigui KC, Boxer LD, Chang HY et al: SIRT6 links histone H3 lysine 9 deacetylation to NF-kappaB-dependent gene expression and organismal life span. Cell 2009, 136(1): 62-74.
- Zhao G, Wang H, Xu C, Wang P, Chen J, Wang P, Sun Z, Su Y, Wang Z, Han L et al: SIRT6 delays cellular senescence by promoting p27Kip1 ubiquitin-proteasome degradation. Aging (Albany NY) 2016, 8(10): 2308-2323.
- Esteller M: Non-coding RNAs in human disease. Nat Rev Genet 2011, 12(12): 861-874.
- Niderla-Bielińska J, Jankowska-Steifer E, Włodarski P: Non-Coding RNAs and Human Diseases: Current Status and Future Perspectives. Int J Mol Sci 2023, 24(14): 11679.
- Zhang QY, Zhou H, Zhou XX, Yu FB, Liu YY, Chen ZY, Ma YQ, Li XL, Tian B: Small non-coding RNAome changes during human chondrocyte senescence as potential epigenetic targets in age-related osteoarthritis. Genomics 2023, 115(2): 110574.
- Wang H, Wang W, Wang J, Zhang L, Luo Y, Tang X: MicroRNA-15a/β1,4-GalT-I axis contributes to cartilage degeneration via NF-κB signaling in osteoarthritis. Clinics (Sao Paulo) 2023, 78: 100254.
- Zhu H, Yan X, Zhang M, Ji F, Wang S: miR-21-5p protects IL-1β-induced human chondrocytes from degradation. J Orthop Surg Res 2019, 14(1): 118.
- Philipot D, Guérit D, Platano D, Chuchana P, Olivotto E, Espinoza F, Dorandeu A, Pers YM, Piette J, Borzi RM et al: p16INK4a and its regulator miR-24 link senescence and chondrocyte terminal differentiation-associated matrix remodeling in osteoarthritis. Arthritis Res Ther 2014, 16(1): R58.
- Liu L, Zhao C, Zhang H, Lu Y, Luo B, Yao Z, Shao Y, Zeng H, Zeng C, Zhang R et al: Asporin regulated by miR-26b-5p mediates chondrocyte senescence and exacerbates osteoarthritis progression via TGF-β1/Smad2 pathway. Rheumatology (Oxford) 2022, 61(6): 2631-2643.
- Shi S, Zhang L, Wang Q, Wang Q, Li D, Sun W, Yi C: Targeting Cartilage miR-195/497 Cluster for Osteoarthritis Treatment Regulates the Circadian Clock. Gerontology 2024, 70(1): 59-75.
- Zhao J, Li C, Qin T, Jin Y, He R, Sun Y, Liu Z, Wu T, Duan C, Cao Y et al: Mechanical overloading-induced miR-325-3p reduction promoted chondrocyte senescence and exacerbated facet joint degeneration. Arthritis Res Ther 2023, 25(1): 54.
- Liu Y, Zhang Z, Lu X, Liu C, Zhang H: Senescence-responsive miR-33-5p promotes chondrocyte senescence and osteoarthritis progression by targeting SIRT6. Int Immunopharmacol 2023, 121: 110506.
- Yan S, Dong W, Li Z, Wei J, Han T, Wang J, Lin F: Metformin regulates chondrocyte senescence and proliferation through microRNA-34a/SIRT1 pathway in osteoarthritis. J Orthop Surg Res 2023, 18(1): 198.
- Zhang W, Hsu P, Zhong B, Guo S, Zhang C, Wang Y, Luo C, Zhan Y, Zhang C: MiR-34a Enhances Chondrocyte Apoptosis, Senescence and Facilitates Development of Osteoarthritis by Targeting DLL1 and Regulating PI3K/AKT Pathway. Cell Physiol Biochem 2018, 48(3): 1304-1316.
- Lu Y, Liu L, Pan J, Luo B, Zeng H, Shao Y, Zhang H, Guan H, Guo D, Zeng C et al: MFG-E8 regulated by miR-99b-5p protects against osteoarthritis by targeting chondrocyte senescence and macrophage reprogramming via the NF-κB pathway. Cell Death Dis 2021, 12(6): 533.
- Qin C, Feng Y, Yin Z, Wang C, Yin R, Li Y, Chen K, Tao T, Zhang K, Jiang Y et al: The PIEZO1/miR-155-5p/GDF6/SMAD2/3 signaling axis is involved in inducing the occurrence and progression of osteoarthritis under excessive mechanical stress. Cell Signal 2024, 118: 111142.
- Kang D, Shin J: Stress-activated miR-204 governs senescent phenotypes of chondrocytes to promote osteoarthritis development. Sci Transl Med 2019, 11(486): eaar6659.
- Ji ML, Li Z, Hu XY, Zhang WT, Zhang HX, Lu J: Dynamic chromatin accessibility tuning by the long noncoding RNA ELDR accelerates chondrocyte senescence and osteoarthritis. Am J Hum Genet 2023, 110(4): 606-624.
- Qian M, Shi Y, Lu W: LINC00707 knockdown inhibits IL-1β-induced apoptosis and extracellular matrix degradation of osteoarthritis chondrocytes by the miR-330-5p/FSHR axis. Immunopharmacol Immunotoxicol 2022, 44(5): 671-681.
- Liu C, Gao J, Su G, Xiang Y, Wan L: MicroRNA-1202 plays a vital role in osteoarthritis via KCNQ1OT1 has-miR-1202-ETS1 regulatory pathway. J Orthop Surg Res 2020, 15(1): 130.
- Lü G, Li L, Wang B, Kuang L: LINC00623/miR-101/HRAS axis modulates IL-1β-mediated ECM degradation, apoptosis and senescence of osteoarthritis chondrocytes. Aging (Albany NY) 2020, 12(4): 3218-3237.
- Ni W, Zhang H, Mei Z, Hongyi Z, Wu Y, Xu W, Ma Y, Yang W, Liang Y, Gu T et al: An inducible long noncoding RNA, LncZFHX2, facilitates DNA repair to mediate osteoarthritis pathology. Redox Biol 2023, 66: 102858.
- Liang Y, Shen L, Ni W, Ding Y, Yang W, Gu T, Zhang C, Yik JHN, Haudenschild DR, Fan S et al: CircGNB1 drives osteoarthritis pathogenesis by inducing oxidative stress in chondrocytes. Clin Transl Med 2023, 13(8): e1358.
- Gong Z, Zhu J, Chen J, Feng F, Zhang H, Zhang Z: CircRREB1 mediates lipid metabolism related senescent phenotypes in chondrocytes through FASN post-translational modifications. Nat Commun 2023, 14(1): 5242.
- Chen F, Wang S, Zeng C, Tang S, Gu H, Wang Z, Li J, Feng P, Zhang Y, Wang P et al: Silencing circSERPINE2 restrains mesenchymal stem cell senescence via the YBX3/PCNA/p21 axis. Cell Mol Life Sci 2023, 80(11): 325.
- Xiao F, Wang C, Peng J, Zhou X, Ma D, Wang Y, Li Y, Chen X, Wang C: Changes in Small Noncoding RNA Expression during Chondrocyte Senescence. Cartilage 2022, 13(3): 19476035221118165.
- Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, Athineos D, Kang TW, Lasitschka F, Andrulis M et al: A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol 2013, 15(8): 978-990.
- Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doorn R, Desmet CJ, Aarden LA, Mooi WJ, Peeper DS: Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 2008, 133(6): 1019-1031.
- Borzì RM, Mazzetti I, Cattini L, Uguccioni M, Baggiolini M, Facchini A: Human chondrocytes express functional chemokine receptors and release matrix-degrading enzymes in response to C-X-C and C-C chemokines. Arthritis Rheum 2000, 43(8): 1734-1741.
- Di Micco R, Krizhanovsky V, Baker D, d’Adda di Fagagna F: Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat Rev Mol Cell Biol 2021, 22(2): 75-95.
- Diekman BO, Sessions GA: Expression of p16(INK)(4a) is a biomarker of chondrocyte aging but does not cause osteoarthritis. Aging Cell 2018, 17(4): e12771.
- Herranz N, Gil J: Mechanisms and functions of cellular senescence. J Clin Invest 2018, 128(4): 1238-1246.
- Liu T, Zhang L, Joo D, Sun SC: NF-κB signaling in inflammation. Signal Transduct Target Ther 2017, 2: 17023.
- Salminen A, Kauppinen A, Kaarniranta K: Emerging role of NF-κB signaling in the induction of senescence-associated secretory phenotype (SASP). Cell Signal 2012, 24(4): 835-845.
- Guo Q, Chen X, Chen J, Zheng G, Xie C, Wu H, Miao Z, Lin Y, Wang X, Gao W et al: STING promotes senescence, apoptosis, and extracellular matrix degradation in osteoarthritis via the NF-κB signaling pathway. Cell Death Dis 2021, 12(1): 13.
- Freund A, Patil CK, Campisi J: p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J 2011, 30(8): 1536-1548.
- Liu Y, Zhang Z, Li J, Chang B, Lin Q, Wang F, Wang W, Zhang H: Piezo1 transforms mechanical stress into pro senescence signals and promotes osteoarthritis severity. Mech Ageing Dev 2023, 216: 111880.
- Laberge RM, Sun Y, Orjalo AV, Patil CK, Freund A, Zhou L, Curran SC, Davalos AR, Wilson-Edell KA, Liu S et al: MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat Cell Biol 2015, 17(8): 1049-1061.
- Yagi M, Endo K, Komori K, Sekiya I: Comparison of the effects of oxidative and inflammatory stresses on rat chondrocyte senescence. Sci Rep 2023, 13(1): 7697.
- Jeon OH, David N, Campisi J, Elisseeff JH: Senescent cells and osteoarthritis: a painful connection. J Clin Invest 2018, 128(4): 1229-1237.
- Chou CH, Jain V, Gibson J, Attarian DE, Haraden CA, Yohn CB, Laberge RM, Gregory S, Kraus VB: Synovial cell cross-talk with cartilage plays a major role in the pathogenesis of osteoarthritis. Sci Rep 2020, 10(1): 10868.
- Orjalo AV, Bhaumik D, Gengler BK, Scott GK, Campisi J: Cell surface-bound IL-1alpha is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc Natl Acad Sci U S A 2009, 106(40): 17031-17036.
- Acosta JC, O'Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, Fumagalli M, Da Costa M, Brown C, Popov N et al: Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 2008, 133(6): 1006-1018.
- Barreto G, Senturk B, Colombo L, Brück O, Neidenbach P, Salzmann G, Zenobi-Wong M, Rottmar M: Lumican is upregulated in osteoarthritis and contributes to TLR4-induced pro-inflammatory activation of cartilage degradation and macrophage polarization. Osteoarthritis Cartilage 2020, 28(1): 92-101.
- Takayama K, Kawakami Y, Lee S, Greco N, Lavasani M, Mifune Y, Cummins JH, Yurube T, Kuroda R, Kurosaka M et al: Involvement of ERCC1 in the pathogenesis of osteoarthritis through the modulation of apoptosis and cellular senescence. J Orthop Res 2014, 32(10): 1326-1332.
- Nelson G, Wordsworth J, Wang C, Jurk D, Lawless C, Martin-Ruiz C, von Zglinicki T: A senescent cell bystander effect: senescence-induced senescence. Aging Cell 2012, 11(2): 345-349.
- Varela-Eirín M, Carpintero-Fernández P, Guitián-Caamaño A, Varela-Vázquez A, García-Yuste A, Sánchez-Temprano A, Bravo-López SB, Yañez-Cabanas J, Fonseca E, Largo R: Extracellular vesicles enriched in connexin 43 promote a senescent phenotype in bone and synovial cells contributing to osteoarthritis progression. Cell Death Dis 2022, 13(8): 681.
- Cao X, Luo P, Huang J, Liang C, He J, Wang Z, Shan D, Peng C, Wu S: Intraarticular senescent chondrocytes impair the cartilage regeneration capacity of mesenchymal stem cells. Stem Cell Res Ther 2019, 10(1): 86.
- Zhou J, Huang J, Li Z, Song Q, Yang Z, Wang L, Meng Q: Identification of aging-related biomarkers and immune infiltration characteristics in osteoarthritis based on bioinformatics analysis and machine learning. Front Immunol 2023, 14: 1168780.
- Felekkis K, Pieri M: Exploring the Feasibility of Circulating miRNAs as Diagnostic and Prognostic Biomarkers in Osteoarthritis: Challenges and Opportunities. Int J Mol Sci 2023, 24(17): 13144.
Asia-Pacific Journal of Surgical & Experimental Pathology
ISSN 2977-5817 (Online)
 Copyright © Asia Pac J Surg Exp & Pathol. This
work is licensed under a Creative Commons AttributionNonCommercial-No Derivatives 4.0 International (CC BY-NC-ND 4.0)
License.
Copyright © Asia Pac J Surg Exp & Pathol. This
work is licensed under a Creative Commons AttributionNonCommercial-No Derivatives 4.0 International (CC BY-NC-ND 4.0)
License.