
 Submit Manuscript
Submit Manuscript
Review Article | Open Access
Role of PD-1/PD-L1 crosstalk on inhibition of T-cell activation and proliferation through blockade of PI3K/Akt/mTOR signaling pathway
Jacob Smith1
1Faculty of Health Sciences, Universidade de Belas, Luanda, Angola.
Correspondence: Jacob Smith (Faculty of Health Sciences, Universidade de Belas, Luanda, Angola; E-mail: jacobsmith.he@yahoo.com).
Asia-Pacific Journal of Surgical & Experimental Pathology 2024, 1: 49-56. https://doi.org/10.32948/ajsep.2024.11.18
Received: 10 Oct 2024 | Accepted: 15 Nov 2024 | Published online: 21 Nov 2024
Key words PD-1/PD-L1 crosstalk, PI3K/Akt/mTOR, signaling pathway, T-cell activation, T-cell proliferation
The PD-1/PD-L1-induced blockade of T-cell proliferation and differentiation is responsible for the immune escape of cancer cells [4]. Therefore, to enhance immune surveillance, activation and proliferation of T-cells and inhibition of the PD-1/PD-L1 crosstalk are breakthroughs of cancer research. T-cells and other hematopoietic cells require glucose uptake and metabolism for survival and function. Glucose transporter 1 (GLUT1) is required for glucose uptake in T-cells, thereby higher expression of GLUT1 is very essential for the activation of T-cells [5]. The PI3K/Akt/mTOR is a crucial mechanism by which T-cell proliferation and differentiation occurs through the upregulation of GLUT1 [6], and the PI3K/Akt/mTOR signaling pathway plays a key role in cytokine-regulated GLUT1 trafficking [6]. Additionally, activation of the PI3K/Akt/mTOR pathway acts as a transducer of CD28 signals to enhance glucose uptake [7]. However, there are numerous underlying mechanisms through which the PD-1/PD-L1 axis suppresses T-cell activation via inhibition of PI3K/Akt/mTOR signaling pathway. During T-cell co-receptors CD3 and CD28-mediated stimulation, phosphatase and tension homolog (PTEN) is phosphorylated by casein kinase 2 (CK2), which stabilizes PTEN and suppresses PTEN phosphatase activity, and this mechanism co-stimulates T-cells [8]. The PD-1/PD-L1 crosstalk abrogates PI3K/Akt/mTOR signaling through increasing PTEN phosphatase activity and decreasing stability of PTEN, leading to inhibition of T-cell activation [9]. Treg cell regulates T-cell development and suppresses activation to avoid autoimmunity. PD-1/PD-L1 facilitates Treg cell development through the blockade of PI3K/Akt/mTOR cascade. PD-1/PD-L1-induced FOXP3 transcription accelerates the Treg cell proliferation by preventing PI3K/Akt/mTOR signaling pathway. It has been reported that premature termination of TCR signaling and inhibition of the PI3K/Akt/mTOR axis increase induction of FOXP3 expression and Treg like gene expression [10]. TGF-β is a potent activator of FOXP3 expression, and it synergizes with PD-1 to inhibit PI3K/Akt/mTOR action. Blockade of PD-L1 impairs peripherally induced Treg (iTreg) cell development, and loss of PD-L1 also reduces the expression of TGF-β, which is critical for the function of iTreg cells [11]. Additionally, PI3K/Akt/mTOR signaling pathway induces the cell cycle transition of T-cells and leads to T-cell proliferation, but the PD-1/PD-L1 axis inhibits the cell cycle transition of T-cells via blockade of PI3K/Akt/mTOR signaling and by downregulation of cell cycle checkpoint proteins, and upregulation of cyclin-dependent kinase inhibitors. SKP2 is a transcription factor that initiates the cell cycle transition of the G1 phase, however, PD-1 suppresses SKP2 transcription by reducing the action of PI3K/Akt/mTOR cascade and MEK/ERK pathway [12]. Moreover, the PD-1/PD-L1 axis alters the metabolic reprogramming of T-cells by abrogating the action of PI3K/Akt/mTOR cascade. PD-1/PD-L1 inhibits GLUT1 expression, blocks glucose uptake in T-cells, and induces β-oxidation of fatty acid to release ATP which provides energy to the T-cells. However, the metabolic process of β-oxidation of fatty acid is fatal for the T-cells because the essential component of the cell membrane is phospholipid which breaks down, and leads to inhibition of T-cell proliferation and breakdown of T-cells [13, 14].
The expression of PD-1 in naïve T-cells is tightly regulated but its expression rapidly proliferates when TCR is activated, and it is the protective mechanism to inhibit excessive activation of immune systems [21]. Pro-inflammatory cytokine IFN-γ is secreted by T-cells and natural killer cells (NK) to enhance neo-antigen presentation by major histocompatibility complex (MHC) on tumor cells [22]. PD-L1 is upregulated by IFN-γ, tumor necrosis factor α (TNFα), interleukin-6 (IL-6) and over-expression of PD-L1 by oncogenic pathways allowing cancer cells to escape immunesurveillance and promote their survival and initiates metastasis by abrogating the immune activity of T-cells [23]. IFN-γ has multiple ways to upregulate PD-L1 for attenuating the action of T-cells, and IFN-γ induces PD-L1 via multiple signaling pathways in different types of cancer. IFN-γ induces PD-L1 upregulation via JAK2/STAT1/IFR-1 signaling pathway in gastric cancer [24] [25]. However, IFN-γ-mediated overexpression of PD-L1 occurs via PI3K/Akt and JAK/STAT3 pathways in lung cancer [26]. IFN-α also acts with TCR signal to regulate PD-1 expression and potentially inhibits T-cell-mediated immune response [27]. IL-6 activates JAK1, and JAK1 phosphorylates Tyr112 of PD-L1, which recruits N-glycosyltransferase STT3A to catalyze PD-L1 glycosylation and regulate stability of PD-L1 [23]. Increased expression of IL-6 is associated with higher expression of PD-L1, and IL-6 also induces expression of PD-1 in activated T-cells [28]. IL-6 and IL-12 additionally act to induce PD-1 expression upon TCR activation and enhances PD-1 transcription via STAT3/STAT4 pathway [29]. IL-6/JAK-mediated protein stability enhances T-cell exhaustion through the synergistic action of T-cell immunoglobulin mucin-3 (Tim-3) and PD-1 on tumor- infiltrating leukocytes including CD8+ T-cells (Figure 1) [30]. The blockade of T-cell functions is correlated with PD-1 and numerous inhibitory receptors including cytotoxic T-lymphocyte-associated antigen 4 (CTLA4), lymphocytes-activation gene 3 (LAG-3), CD160, and 2B4 [31].
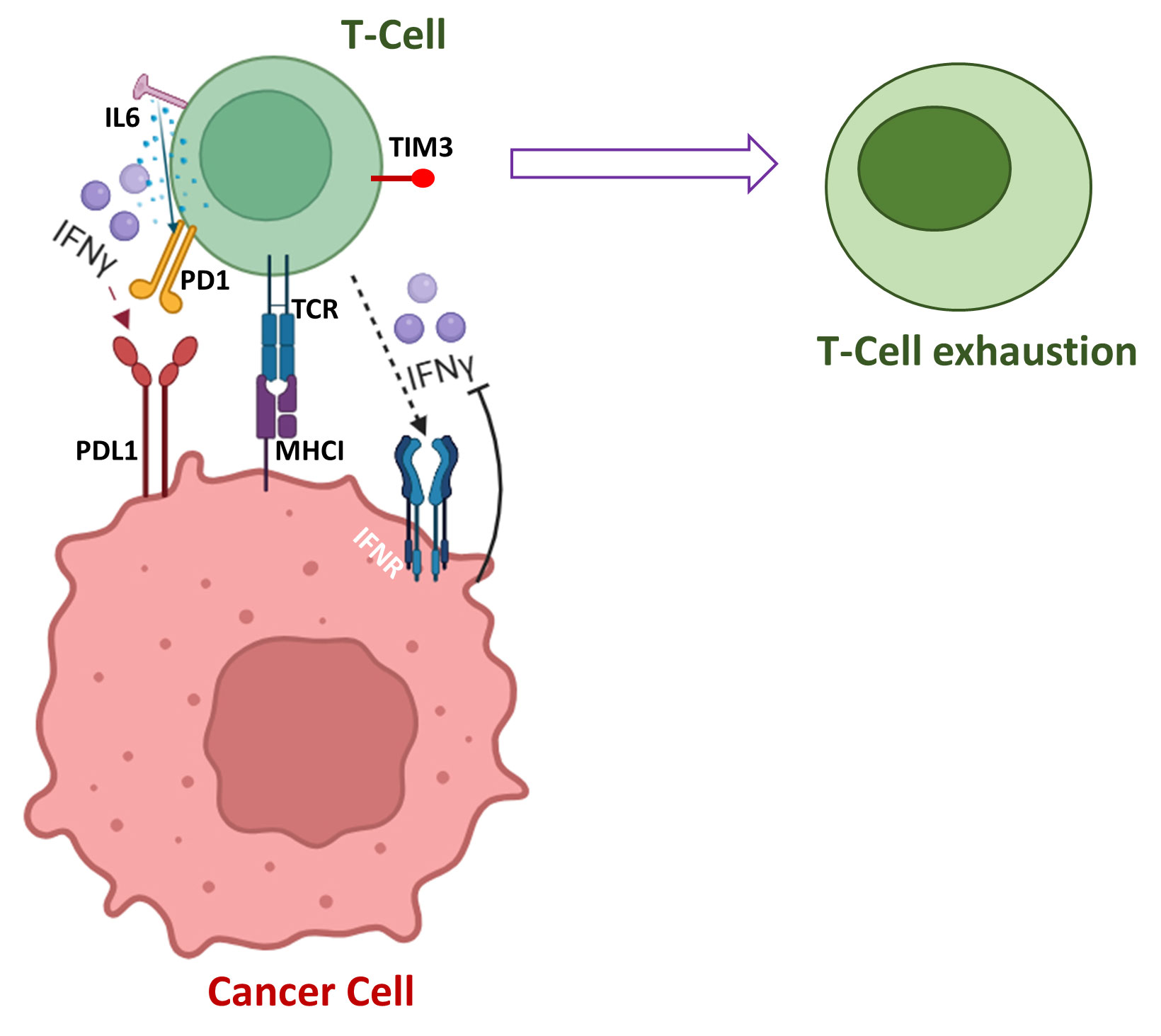 Figure 1. Numerous pathways of T-cell exhaustion.
Figure 1. Numerous pathways of T-cell exhaustion.
PI3K has four different classes, among them IA and IB classes of PI3K was studied extensively on T-cells [55]. It was revealed that besides the crucial function of thymic development of nTreg and iTreg for the suppression of excessive immune response to avoid autoimmunity, TGF-β also initiates a specific type of immune response by co-activation of some cytokines. TGF-β selectively mediates Akt phosphorylation exclusively at the ser473 domain but not at the Thr308 domain in a class IA PI3K-dependent manner, leading to the inhibition of Foxo transcription factors and prevention of the differentiation of iTreg cells. TGF-β-induced Akt phosphorylation reduces the differentiation of iTreg, while the elimination of the p85α subunit, class IA PI3K enhances iTreg differentiation [56].
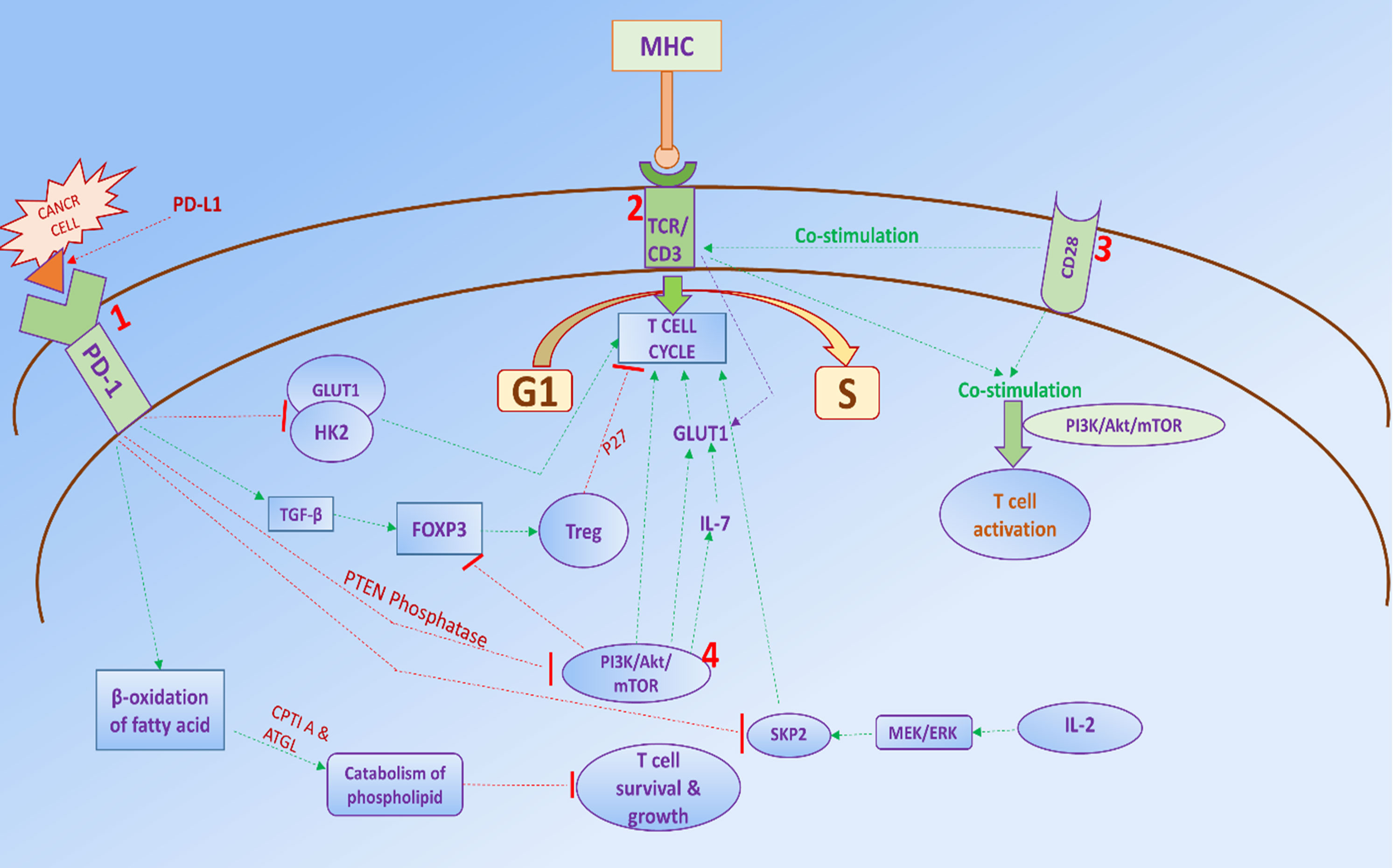 Figure 2. (1) After ligation of the PD-1 receptor with PD-L1 ligand, it inhibits PI3K/Akt/mTOR signaling through numerous mechanisms, such as enhancing the activity of PTEN phosphatase. It also prevents cell cycle transition of T-cells by inducing Treg cells with accelerating the action of TGF-B by reducing SKP2 expression through upregulation of GLUT1 and HK2 and inhibiting PI3K/Akt/mTOR signaling. In addition, Treg induces cell cycle arrest by increasing the expression of CDK inhibitor p27. PD-1/PD-L1 also reduces glucose uptake by T-cells and triggers B-oxidation of fatty acid, and catabolism of phospholipid occurs, leading to a reduction of T-cell survival and growth. (2) After co-stimulation of the TCR/CD3 complex, activates T-cell through the PI3K/Akt/mTOR axis. And increase glucose uptake of T-cell via upregulating GLUT1, thus initiating cell cycle transition of T-cell. (3) CD28 plays a crucial role in stimulating TCR/CD3 complex and activating T-cells via enhancing glucose uptake. (4) PI3K/Akt/mTOR pathway accelerates cell cycle of T-cells via downstream expression of IL-7 and GLUT1; it also reduces Treg development through downregulation FOXP3 expression.
Figure 2. (1) After ligation of the PD-1 receptor with PD-L1 ligand, it inhibits PI3K/Akt/mTOR signaling through numerous mechanisms, such as enhancing the activity of PTEN phosphatase. It also prevents cell cycle transition of T-cells by inducing Treg cells with accelerating the action of TGF-B by reducing SKP2 expression through upregulation of GLUT1 and HK2 and inhibiting PI3K/Akt/mTOR signaling. In addition, Treg induces cell cycle arrest by increasing the expression of CDK inhibitor p27. PD-1/PD-L1 also reduces glucose uptake by T-cells and triggers B-oxidation of fatty acid, and catabolism of phospholipid occurs, leading to a reduction of T-cell survival and growth. (2) After co-stimulation of the TCR/CD3 complex, activates T-cell through the PI3K/Akt/mTOR axis. And increase glucose uptake of T-cell via upregulating GLUT1, thus initiating cell cycle transition of T-cell. (3) CD28 plays a crucial role in stimulating TCR/CD3 complex and activating T-cells via enhancing glucose uptake. (4) PI3K/Akt/mTOR pathway accelerates cell cycle of T-cells via downstream expression of IL-7 and GLUT1; it also reduces Treg development through downregulation FOXP3 expression.
The PI3K/Akt/mTOR signaling pathway is one of the key regulators of T-cells and not only governs the activation, differentiation, survival, and proliferation of T-cells, but also modulates other cytokines, interleukins and transcription factors which directly or indirectly sharpen the immune mechanism through modulating the action of T-cells. Autoimmunity is the state where the immune system reacts against its own normal components, healthy cells, and tissue, and it could be very harmful when its own normal cells start destroying by the autoimmune response. To avoid autoimmunity, PD-1/PD-L1 axis suppresses activation of T-cells via blockade of the PI3K/Akt/mTOR signaling pathway, which negatively impacts on tight regulation of the immune system, and weakens the immune surveillance. The cancer cells get an advantage of immune escape mechanism by which cancer cells proliferate uncontrolled manner. Thereby, for the activation and growth of T-cells, restoring the action of PI3K/Akt/mTOR cascade through inhibition of PD1/PD-L1 crosstalk is a breakthrough in cancer research.
Acknowledgments
No applicable.
Ethics approval
No applicable.
Data availability
The Data will be available upon request.
Funding
None.
Authors’ contribution
JS contributed to the conception, design, writing of this review article and submitted the final version of the manuscript.
Competing interests
None.
- Donátová K, Nováková E, Šupolíková M: Immunotherapy for cancer treatment. Klin Onkol 2022, 35(4): 284-289.
- Keir ME, Butte MJ, Freeman GJ, Sharpe AH: PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol 2008, 26: 677-704.
- Francisco LM, Sage PT, Sharpe AH: The PD-1 pathway in tolerance and autoimmunity. Immunol Rev 2010, 236: 219-242.
- Liu J, Chen Z, Li Y, Zhao W, Wu J, Zhang Z: PD-1/PD-L1 Checkpoint Inhibitors in Tumor Immunotherapy. Front Pharmacol 2021, 12: 731798.
- Maciver NJ, Jacobs SR, Wieman HL, Wofford JA, Coloff JL, Rathmell JC: Glucose metabolism in lymphocytes is a regulated process with significant effects on immune cell function and survival. J Leukoc Biol 2008, 84(4): 949-957.
- Wieman HL, Wofford JA, Rathmell JC: Cytokine stimulation promotes glucose uptake via phosphatidylinositol-3 kinase/Akt regulation of Glut1 activity and trafficking. Mol Biol Cell 2007, 18(4): 1437-1446.
- Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR, Elstrom RL, June CH, Thompson CB: The CD28 signaling pathway regulates glucose metabolism. Immunity 2002, 16(6): 769-777.
- Patsoukis N, Li L, Sari D, Petkova V, Boussiotis VA: PD-1 increases PTEN phosphatase activity while decreasing PTEN protein stability by inhibiting casein kinase 2. Mol Cell Biol 2013, 33(16): 3091-3098.
- Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, Sharpe AH: PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med 2009, 206(13): 3015-3029.
- Sauer S, Bruno L, Hertweck A, Finlay D, Leleu M, Spivakov M, Knight ZA, Cobb BS, Cantrell D, O'Connor E et al: T cell receptor signaling controls Foxp3 expression via PI3K, Akt, and mTOR. Proc Natl Acad Sci U S A 2008, 105(22): 7797-7802.
- Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, Sharpe AH: PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med 2009, 206(13): 3015-3029.
- Patsoukis N, Brown J, Petkova V, Liu F, Li L, Boussiotis VA: Selective effects of PD-1 on Akt and Ras pathways regulate molecular components of the cell cycle and inhibit T cell proliferation. Sci signal 2012, 5(230): ra46-ra46.
- Deberardinis RJ, Lum JJ, Thompson CB: Phosphatidylinositol 3-kinase-dependent modulation of carnitine palmitoyltransferase 1A expression regulates lipid metabolism during hematopoietic cell growth. J Biol Chem 2006, 281(49): 37372-37380.
- Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, Karoly ED, Freeman GJ, Petkova V, Seth P et al: PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat Commun 2015, 6: 6692.
- Jiang X, Wang J, Deng X, Xiong F, Ge J, Xiang B, Wu X, Ma J, Zhou M, Li X et al: Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol Cancer 2019, 18(1): 10.
- Wei W, Ayad NG, Wan Y, Zhang GJ, Kirschner MW, Kaelin WG, Jr: Degradation of the SCF component Skp2 in cell-cycle phase G1 by the anaphase-promoting complex. Nature 2004, 428(6979): 194-198.
- Zhang X, Schwartz JC, Guo X, Bhatia S, Cao E, Lorenz M, Cammer M, Chen L, Zhang ZY, Edidin MA et al: Structural and functional analysis of the costimulatory receptor programmed death-1. Immunity 2004, 20(3): 337-347.
- Ahmadzadeh M, Johnson LA, Heemskerk B, Wunderlich JR, Dudley ME, White DE, Rosenberg SA: Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 2009, 114(8): 1537-1544.
- Greenwald RJ, Freeman GJ, Sharpe AH: The B7 family revisited. Annu Rev Immunol 2005, 23: 515-548.
- Hofmeyer KA, Jeon H, Zang X: The PD-1/PD-L1 (B7-H1) pathway in chronic infection-induced cytotoxic T lymphocyte exhaustion. J Biomed Biotechnol 2011, 2011: 451694.
- Agata Y, Kawasaki A, Nishimura H, Ishida Y, Tsubata T, Yagita H, Honjo T: Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int Immunol 1996, 8(5): 765-772.
- Cha JH, Chan LC, Li CW, Hsu JL, Hung MC: Mechanisms Controlling PD-L1 Expression in Cancer. Mol Cell 2019, 76(3): 359-370.
- Chan LC, Li CW, Xia W, Hsu JM, Lee HH, Cha JH, Wang HL, Yang WH, Yen EY, Chang WC et al: IL-6/JAK1 pathway drives PD-L1 Y112 phosphorylation to promote cancer immune evasion. J Clin Invest 2019, 129(8): 3324-3338.
- Mimura K, Teh JL, Okayama H, Shiraishi K, Kua LF, Koh V, Smoot DT, Ashktorab H, Oike T, Suzuki Y et al: PD-L1 expression is mainly regulated by interferon gamma associated with JAK-STAT pathway in gastric cancer. Cancer Sci 2018, 109(1): 43-53.
- Moon JW, Kong SK, Kim BS, Kim HJ, Lim H, Noh K, Kim Y, Choi JW, Lee JH, Kim YS: IFNγ induces PD-L1 overexpression by JAK2/STAT1/IRF-1 signaling in EBV-positive gastric carcinoma. Sci Rep 2017, 7(1): 17810.
- Zhang X, Zeng Y, Qu Q, Zhu J, Liu Z, Ning W, Zeng H, Zhang N, Du W, Chen C et al: PD-L1 induced by IFN-γ from tumor-associated macrophages via the JAK/STAT3 and PI3K/AKT signaling pathways promoted progression of lung cancer. Int J Clin Oncol 2017, 22(6): 1026-1033.
- Terawaki S, Chikuma S, Shibayama S, Hayashi T, Yoshida T, Okazaki T, Honjo T: IFN-α directly promotes programmed cell death-1 transcription and limits the duration of T cell-mediated immunity. J Immunol 2011, 186(5): 2772-2779.
- Johnson DE, O'Keefe RA, Grandis JR: Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat Rev Clin Oncol 2018, 15(4): 234-248.
- Austin JW, Lu P, Majumder P, Ahmed R, Boss JM: STAT3, STAT4, NFATc1, and CTCF regulate PD-1 through multiple novel regulatory regions in murine T cells. J Immunol 2014, 192(10): 4876-4886.
- Patsoukis N, Brown J, Petkova V, Liu F, Li L, Boussiotis VA: Selective effects of PD-1 on Akt and Ras pathways regulate molecular components of the cell cycle and inhibit T cell proliferation. Sci Signal 2012, 5(230): ra46.
- Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, Betts MR, Freeman GJ, Vignali DA, Wherry EJ: Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol 2009, 10(1): 29-37.
- Jacobs SR, Herman CE, Maciver NJ, Wofford JA, Wieman HL, Hammen JJ, Rathmell JC: Glucose uptake is limiting in T cell activation and requires CD28-mediated Akt-dependent and independent pathways. J Immunol 2008, 180(7): 4476-4486.
- Han JM, Patterson SJ, Levings MK: The Role of the PI3K Signaling Pathway in CD4(+) T Cell Differentiation and Function. Front Immunol 2012, 3: 245.
- Huang J, Manning BD: The TSC1-TSC2 complex: a molecular switchboard controlling cell growth. Biochem J 2008, 412(2): 179-190.
- Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, Sullivan SA, Nichols AG, Rathmell JC: Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol 2011, 186(6): 3299-3303.
- Cornish GH, Sinclair LV, Cantrell DA: Differential regulation of T-cell growth by IL-2 and IL-15. Blood 2006, 108(2): 600-608.
- Schluns KS, Lefrançois L: Cytokine control of memory T-cell development and survival. Nat Rev Immunol 2003, 3(4): 269-279.
- Macintyre AN, Finlay D, Preston G, Sinclair LV, Waugh CM, Tamas P, Feijoo C, Okkenhaug K, Cantrell DA: Protein kinase B controls transcriptional programs that direct cytotoxic T cell fate but is dispensable for T cell metabolism. Immunity 2011, 34(2): 224-236.
- Kim EH, Sullivan JA, Plisch EH, Tejera MM, Jatzek A, Choi KY, Suresh M: Signal integration by Akt regulates CD8 T cell effector and memory differentiation. J Immunol 2012, 188(9): 4305-4314.
- Chen CC, Jeon SM, Bhaskar PT, Nogueira V, Sundararajan D, Tonic I, Park Y, Hay N: FoxOs inhibit mTORC1 and activate Akt by inducing the expression of Sestrin3 and Rictor. Dev Cell 2010, 18(4): 592-604.
- Park Y, Jin H-S, Lopez J, Elly C, Kim G, Murai M, Kronenberg M, Liu Y-C: TSC1 regulates the balance between effector and regulatory T cells. Journal Clin Invest 2013, 123(12): 5165-5178.
- Semenza GL: Hypoxia-inducible factor 1 (HIF-1) pathway. Sci STKE 2007, 2007(407): cm8.
- Delgoffe GM, Kole TP, Cotter RJ, Powell JD: Enhanced interaction between Hsp90 and raptor regulates mTOR signaling upon T cell activation. Mol Immunol 2009, 46(13): 2694-2698.
- Wang X, Huang H, Young KH: The PTEN tumor suppressor gene and its role in lymphoma pathogenesis. Aging 2015, 7(12): 1032-1049.
- Riley JL: PD-1 signaling in primary T cells. Immunol Rev 2009, 229(1): 114-125.
- Torres J, Pulido R: The tumor suppressor PTEN is phosphorylated by the protein kinase CK2 at its C terminus. Implications for PTEN stability to proteasome-mediated degradation. J Biol Chem 2001, 276(2): 993-998.
- Cai J, Wang D, Zhang G, Guo X: The Role Of PD-1/PD-L1 Axis In Treg Development And Function: Implications For Cancer Immunotherapy. Onco Targets Ther 2019, 12: 8437-8445.
- Malchow S, Leventhal DS, Nishi S, Fischer BI, Shen L, Paner GP, Amit AS, Kang C, Geddes JE, Allison JP et al: Aire-dependent thymic development of tumor-associated regulatory T cells. Science 2013, 339(6124): 1219-1224.
- Pompura SL, Dominguez-Villar M: The PI3K/AKT signaling pathway in regulatory T-cell development, stability, and function. J Leukoc Biol 2018, https://doi.org/10.1002/jlb.2mir0817-349r. Epub ahead of print.
- Haxhinasto S, Mathis D, Benoist C: The AKT-mTOR axis regulates de novo differentiation of CD4+Foxp3+ cells. J Exp Med 2008, 205(3): 565-574.
- Fontenot JD, Rasmussen JP, Williams LM, Dooley JL, Farr AG, Rudensky AY: Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity 2005, 22(3): 329-341.
- Que Y, Xiao W, Guan Y-x, Liang Y, Yan S-m, Chen H-y, Li Q-q, Xu B-s, Zhou Z-w, Zhang X: PD-L1 Expression Is Associated with FOXP3+ Regulatory T-Cell Infiltration of Soft Tissue Sarcoma and Poor Patient Prognosis. J Cancer 2017, 8(11): 2018-2025.
- Beswick EJ, Pinchuk IV, Das S, Powell DW, Reyes VE: Expression of the Programmed Death Ligand 1, B7-H1, on Gastric Epithelial Cells after Helicobacter pylori Exposure Promotes Development of CD4+ CD25+FoxP3+ Regulatory T Cells. Infect Immun 2007, 75(9): 4334.
- Lohr J, Knoechel B, Abbas AK: Regulatory T cells in the periphery. Immunol Rev 2006, 212(1): 149-162.
- Han JM, Patterson SJ, Levings MK: The Role of the PI3K Signaling Pathway in CD4(+) T Cell Differentiation and Function. Front Immunol 2012, 3: 245-245.
- Kurebayashi Y, Baba Y, Minowa A, Nadya NA, Azuma M, Yoshimura A, Koyasu S, Nagai S: TGF-β-induced phosphorylation of Akt and Foxo transcription factors negatively regulates induced regulatory T cell differentiation. Biochem Biophys Res Commun 2016, 480(1): 114-119.
- Turner BC, Tonks NK, Rapp UR, Reed JC: Interleukin 2 regulates Raf-1 kinase activity through a tyrosine phosphorylation-dependent mechanism in a T-cell line. Proc Natl Acad Sci U S A 1993, 90(12): 5544-5548.
- Remillard B, Petrillo R, Maslinski W, Tsudo M, Strom TB, Cantley L, Varticovski L: Interleukin-2 receptor regulates activation of phosphatidylinositol 3-kinase. J Biol Chem 1991, 266(22): 14167-14170.
- Fry TJ, Connick E, Falloon J, Lederman MM, Liewehr DJ, Spritzler J, Steinberg SM, Wood LV, Yarchoan R, Zuckerman J et al: A potential role for interleukin-7 in T-cell homeostasis. Blood 2001, 97(10): 2983-2990.
- Swainson L, Kinet S, Mongellaz C, Sourisseau M, Henriques T, Taylor N: IL-7-induced proliferation of recent thymic emigrants requires activation of the PI3K pathway. Blood 2007, 109(3): 1034-1042.
- Pallard C, Stegmann AP, van Kleffens T, Smart F, Venkitaraman A, Spits H: Distinct roles of the phosphatidylinositol 3-kinase and STAT5 pathways in IL-7-mediated development of human thymocyte precursors. Immunity 1999, 10(5): 525-535.
- Barata JT, Silva A, Brandao JG, Nadler LM, Cardoso AA, Boussiotis VA: Activation of PI3K is indispensable for interleukin 7-mediated viability, proliferation, glucose use, and growth of T cell acute lymphoblastic leukemia cells. J Exp Med 2004, 200(5): 659-669.
- Phillips R, Ager A: Activation of pertussis toxin-sensitive CXCL12 (SDF-1) receptors mediates transendothelial migration of T lymphocytes across lymph node high endothelial cells. Eur J Immunol 2002, 32(3): 837-847.
- Finlay D, Cantrell D: Phosphoinositide 3-kinase and the mammalian target of rapamycin pathways control T cell migration. Ann N Y Acad Sci 2010, 1183: 149-157.
- Schmelzle T, Hall MN: TOR, a central controller of cell growth. Cell 2000, 103(2): 253-262.
Asia-Pacific Journal of Surgical & Experimental Pathology
ISSN 2977-5817 (Online)
 Copyright © Asia Pac J Surg Exp & Pathol. This
work is licensed under a Creative Commons AttributionNonCommercial-No Derivatives 4.0 International (CC BY-NC-ND 4.0)
License.
Copyright © Asia Pac J Surg Exp & Pathol. This
work is licensed under a Creative Commons AttributionNonCommercial-No Derivatives 4.0 International (CC BY-NC-ND 4.0)
License.